CHAPITRE I : INTRODUCTION
I. ANATOMIE
A. ANATOMIE MACROSCOPIQUE
Le foie est irrigué par une double circulation sanguine :
·
veineuse qui, via la veine porte, y amène le sang récolté
par les vaisseaux intestinaux (veines mésentériques) et coeliaques.
·
artérielle par l’artère hépatique, branche du tronc
coeliaque.
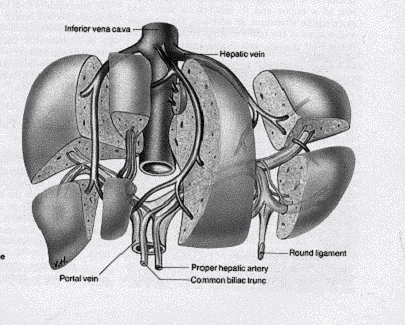
Le sang est drainé par les veines sus-hépatiques qui résultent de la fusion des
veines sublobulaires.
Figure 1
La circulation de bile s’effectue par les canaux biliaires qui forment dans le foie un réseau arborescent parallèle à la circulation sanguine.
L’anatomie chirurgicale du foie divise l’organe en 8 segments. Le lobe gauche comprend les segments II et III. Le foie gauche comprend en outre les segments I et IV. Il y a donc une différence entre l’hépatectomie droite ou gauche (qui est basée sur la notion de foie droit et gauche) et la lobectomie hépatique droite ou gauche (qui est basée sur la notion de lobe droit ou gauche).
B. ANATOMIE MICROSCOPIQUE
Il faut distinguer l’acinus hépatique et le lobule hépatique.
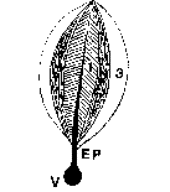 L’unité morphologique
élémentaire est l’acinus de Rappaport
qui est construit autour de la triade portale (artère, veine porte, canal
biliaire) et forme autour de cette dernière une petite masse de forme analogue
à une petite baie. On y distingue classiquement 3 zones (1,2,3) dont
l’oxygénation est différente. Le sang de la branche veineuse se distribue dans
l’acinus. Le sang artériel (à faible débit) arrive sous forte pression et se
mêle au sang veineux au niveau des sinusoïdes hépatiques par un réseau de
shunts, entraînant un effet de chasse qui favorise la circulation. Le sang est mieux oxygéné au centre de l’acinus
(zone 1) qu’en périphérie zone 3), ce qui explique la répartition inégale de
certaines lésions parenchymateuses. Le drainage de l’acinus se fait en
périphérie par le réseau des veines collectrices qualifiées de veineuses
centro-lobulaires.
L’unité morphologique
élémentaire est l’acinus de Rappaport
qui est construit autour de la triade portale (artère, veine porte, canal
biliaire) et forme autour de cette dernière une petite masse de forme analogue
à une petite baie. On y distingue classiquement 3 zones (1,2,3) dont
l’oxygénation est différente. Le sang de la branche veineuse se distribue dans
l’acinus. Le sang artériel (à faible débit) arrive sous forte pression et se
mêle au sang veineux au niveau des sinusoïdes hépatiques par un réseau de
shunts, entraînant un effet de chasse qui favorise la circulation. Le sang est mieux oxygéné au centre de l’acinus
(zone 1) qu’en périphérie zone 3), ce qui explique la répartition inégale de
certaines lésions parenchymateuses. Le drainage de l’acinus se fait en
périphérie par le réseau des veines collectrices qualifiées de veineuses
centro-lobulaires.
Figure 2
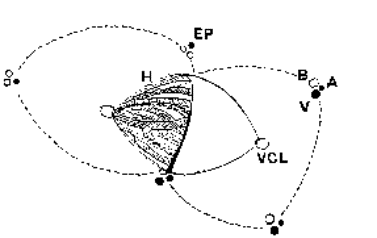
Figure 3
Le lobule hépatique (en pointillé) est axé sur la veine centro-lobulaire (VCL). Il est délimité en périphérie par les espaces portes (EP). Le lobule hépatique ne constitue pas une unité fonctionnelle mais doit être considéré comme une subvision anatomique du foie.
A l’intérieur de l’acinus, les hépatocytes (CH) se disposent en un réseau complexe de cloisons, perforés par les sinusoïdes et les canaux biliaires (CB) de drainage. En coupe ces cloisons se présentent sous forme de lames unistratifiées séparées les unes des autres par les sinusoïdes dont la structure est complexe. Leur endothélium (CE) est largement fenêtré, il ne repose sur aucune membrane basale. Il est séparé des hépatocytes par l’espace de Disse (ED) dans lequel on trouve des fibroblastes, des neurones et des cellules chargées de stocker de la vitamine A (cellules de ITO) (CI). Des cellules macrophagiques nombreuses (cellules de Kupffer) (CK) accolées à l’endothélium flottent dans la lumière du sinusoïde. Elles sont douées d’une importante capacité de phagocytose.

Figure 4
II. FONCTIONS DU FOIE ET SES ALTERATIONS
A. LES FONCTIONS METABOLIQUES
Le foie joue un rôle absolument central dans le métabolisme des trois nutriments de base (protéines, lipides et hydrates de carbone), ainsi que de la plupart des vitamines et de certains oligo-éléments. Ce rôle est assuré par la captation et la conversion de molécules et de particules (provenant de l’absorption intestinale et du métabolisme du tractus digestif et apportées par la veine porte ou mobilisées à partir des tissus périphériques - et amenées par l’artère hépatique), ainsi que par la production et la sécrétion de nouvelles molécules et particules.
1. Métabolisme des protéines
a. Conditions normales (rappel)
Le foie joue un rôle central dans le métabolisme des acides aminés et des protéines. En effet, cet organe prend en charge la synthèse de la majeure partie des protéines circulantes, transforme les acides aminés en substrats énergétiques (glucose et corps cétoniques) et assure l’épuration de l’azote excédentaire.
(1) Synthèse et sécrétion des protéines
Le foie synthétise et sécrète la plupart des protéines fonctionnelles (ou protéines viscérales) présentes dans le sang : l’albumine (transport d’hormones, d’acides gras libres, du calcium,…), la transferrine (transport du fer), la céruloplasmine (transport du cuivre), la transthyrétine ou préalbumine (transport de la thyroxine), la « rétino-binding protein » ou RBP (transport de la vitamine A), des facteurs de coagulation, etc.
La synthèse de ces protéines est dépendante de la disponibilité en précurseurs (les acides aminés) et en zinc. Cette synthèse est réprimée par des médiateurs (cytokines) synthétisés notamment par les cellules du système réticulo-endothélial, qui induisent l’activation de l’ARN d’autres protéines caractéristiques de la réaction inflammatoire.
(2)
Formation de
substrats énergétiques
L’oxydation directe de certains acides aminés conduit à la génération d’énergie, mais ce processus est quantitativement (et qualitativement) limité. En fait, les acides aminés constituent des substrats énergétiques essentiellement au travers de leur transformation en glucose, et dans une bien moindre mesure en corps cétoniques.
On appelle gluconéogenèse la synthèse de glucose à partir de précurseurs non glucidiques. En théorie, la plupart des acides aminés sont gluconéogéniques. En pratique, ce rôle est rempli surtout par l’alanine (ainsi que par la proline et la glycine). D’un point de vue énergétique, la gluconéogenèse à partir d’acides aminés est un processus peu rentable puisque 1,72 g d’acides aminés est sacrifié pour produire 1 g de glucose. Chez l’homme sain, la gluconéogenèse est heureusement peu mise à contribution puisqu’en période interprandiale la glycogénolyse fournit l’essentiel du glucose nécessaire aux tissus extra-hépatiques.
La cétogénèse (ou production de corps cétoniques) est théoriquement possible à partir de certains acides aminés ramifiés, mais ce processus est quantitativement très limité chez l’homme. Par contre, le foie transforme en corps cétoniques les acides a-cétoniques produits dans les tissus périphériques à partir d’acides aminés ramifiés.
b. Conditions pathologiques
(1) Synthèses protéiques
Des atteintes importantes du parenchyme hépatique vont avoir des conséquences fonctionnelles, telles qu’une diminution de synthèse des protéines viscérales, et, à terme par une réduction de production des facteurs de coagulation. Cette dernière complication peut également se rencontrer dans les pathologies cholostatique et est alors consécutive à une malabsorption de vitamines liposolubles (notamment la vitamine K).
Lors d’états de dénutrition, la diminution d’apports
d’acides aminés à partir du tube digestif entraîne une diminution de la
synthèse des protéines viscérales par le foie. La concentration plasmatique des
protéines diminue en fonction de leur demi-vie. Ainsi, la concentration
d’albumine (1/2 vie : 20 j) est un bon marqueur de la dénutrition chronique,
compte tenu de son importance quantitative dans le plasma. Par contre, à court
terme, les modifications de concentrations d’albumine reflètent surtout des
changements de son espace de distribution ; dans ces conditions, la RBP (1/2
vie : 12 h) et la préalbumine (1/2 vie : 48 h) représenteront de bien meilleurs
marqueurs de la dénutrition aiguë ; notons cependant que la concentration de
ces protéines, surtout de la RBP, est augmentée en cas d’altération de la
fonction rénale. La concentration de transferrine (1/2 vie : 7 j) est également
utilisée comme paramètre de l’état nutritionnel, mais elle est influencée par la
concentration de fer.
Dans les états inflammatoires (aigus ou chroniques)
après un traumatisme et au cours de phénomènes septiques, la production de
cytokines inflammatoires (interleukine ou IL 6, IL 1 et « tumour necrosis
factor » ou TNFa) induit une diminution de
synthèse des protéines viscérales, au profit de protéines dites de phase
inflammatoire, comme la « C reactive protein » ou CRP, l’a-1glycoprotéine, l’a-1 antirypsine, l’haptoglobine, le
fibrinogène, etc. Ces protéines possèdent des propriétés utiles pour la réponse
contre l’agression, par exemple l’opsonisation.
(2) Gluconéogenèse
La gluconéogenèse à partir d’acides aminés est utile
pour fournir le glucose indispensable au métabolisme énergétique de certains
tissus spécifiques, après l’épuisement des réserves hépatiques de glycogenèse,
soit après un jeûne prolongé sur plus de 24 à 48 h. Ce processus peut être
perturbé au cours de dommages hépatiques très sévères, comme l’hépatite
fulminante.
Dans les états de dénutrition chronique survenant en
dehors d’agressions, l’importance quantitative du la gluconéogenèse tend à
s’atténuer avec le temps, en raison de la mise en route de mécanismes
d’épargne, comme la diminution des dépenses
énergétiques et l’utilisation de corps cétoniques. Ces derniers mécanisme
n’interviennent pas dans les états d’agression, où les besoins énergétiques
sont nettement accrus (et notamment les besoins en glucose au niveau des tissus
de cicatrisation) et où la cétogenèse est inhibée. Dans ces circonstances, la
gluconéogenèse est véritablement emballée, et au contraire de ce qui est
observé dans les situations courantes, les perfusions de glucose (même en
quantité élevée, soit > 200 j) ne parviennent plus à l’inhiber. La
mobilisation d’acides aminés par protéolyse musculaire est stimulée, non
seulement par les besoins en glucose à couvrir par la gluconéogenèse mais
également par les besoins accrus en glutamine au niveau de la muqueuse
intestinale et des cellules immunitaires, ce métabolisme libérant de grandes
quantités d’alanine.
(3) Altérations du métabolisme des protéines et
des acides aminés liés à des pathologies hépatiques
La cirrhose
est souvent associée à un état de dénutrition protéique. Dans cette pathologie,
on observe souvent une altération du profil des acides aminés plasmatiques,
caractérisée par une élévation des acides aminés aromatiques (phénylalanine,
tyrosine, tryptophane) et une diminution des ramifiés (valine, leucine,
isoleucine, lysine).
Une insuffisance
hépatocellulaire sévère est toujours associée à une production réduite des
protéines viscérales, et les états les plus graves à une synthèse diminuée des
facteurs de coagulation. Ceci explique l’importance de maintenir des apports
protéiques normaux
(0,75-1g/kg de poids) aux malades, à l’exception des
états d’encéphalopathies hépatiques. Dans ces cas, on s’efforce de diminuer la
production de dérivés protidiques neurotoxiques en limitant drastiquement les
apports protéiques (20 à 40 g/j), et de réduire leur absorption en diminuant le
pH colique (par des fibres ou du lactulose).
2. Métabolisme des hydrates de carbone
a. Conditions normales (rappel)
Ce rappel sera succinct. Le métabolisme hépatique
des hydrates de carbone consiste en la formation et le dépôt de glycogène
pendant la période postprandiale et sa mobilisation pendant les périodes
interprandiales. Dans les conditions normales, ces mécanismes, qui sont régulés
par l’équilibre entre l’insuline d’une part et le glucagon et l’adrénaline
d’autre part, règlent de façon très précise l’homéostasie glycémique. Comme
mentionné plus haut, le processus du gluconéogenèse à partir des substrats non
glucidiques (alanine, lactate, pyruvate, glycérol) reste alors limité. La
quantité de glycogène déposé ne dépasse normalement pas 5% du poids du foie, et
l’excédent de substrats glucidiques est converti en acides gras sous l’action
de la lipogenèse.
b. Conditions pathologiques
Nous ne considérerons pas ici les pathologies liées
à un déficit enzymatique congénital : glycogénoses, galactosémie, intolérance
au fructose, ...
L’altération du métabolisme glucidique la plus
fréquemment rencontrée dans les hépatopathies consiste en la résistance à
l’action de l’insuline et l’état de diabète des malades cirrhotiques, d’où
l’utilité d’enrichir leur alimentation en hydrates de carbone complexes et en
fibres.
Une complication plus rare, mais redoutable est
l’hypoglycémie des malades présentant une insuffisance hépatocellulaire aiguë
(cf. plus loin l’hépatite alcoolique grave).
Notons encore que les malades en état d’agression
présentent une résistance à convertir un excès d’apports glucidiques en
graisses et qu’une alimentation hypercalorique peut entraîner chez eux une
accumulation de glycogène (et d’eau), provoquant ainsi une hépatomégalie et une
altération des fonctions du foie.
3. Métabolisme des lipides
a. Conditions normales (rappel)
Le foie se trouve également jouer un rôle très
central dans le métabolisme des lipides et des lipoprotéines. C’est en effet au
niveau hépatique que s’effectue l’essentiel de la lipogenèse (conversion de
glucides excédentaires en acides gras et leur estérification en triglycérides),
de la cholestérogenèse, et de la cétogenèse.
Par ailleurs, le foie fait partir des tissus
spécialisés ayant acquis la capacité d’assurer l’élongation et la désaturation
des acides gras essentiels, transformant ainsi les acides à 18 atomes de
carbone des végétaux (terrestres et marins) en dérivés polyinsaturés.
Le parenchyme hépatique intervient aussi largement
dans le métabolisme des lipoprotéines, en produisant des enzymes-clés (comme la
lipase hépatique, la « lecithin cholesterol acyltransferase » ou
LCAT, et la « cholesteryl ester transfer protein » ou CEPT. Il est
prioritairement impliqué dans la captation des particules résiduelles provenant
du métabolisme périphérique des chylomicrons intestinaux. De plus, c’est par
les hépatocytes qu’est produite l’apoprotéine B-100 qui est incorporée à la
surface des lipoprotéines à très faible densité ou « VLDL » et c’est
au niveau du foie qu’intervient la captation des lipoprotéines à densité
intermédiaire ou « IDL », ou leur conversion en lipoprotéines à
faible densité ou « LDL », ainsi que la captations prioritaire de
celles-ci. Le foie participe, avec l’intestin, à la formation et la sécrétion
des lipoprotéines à haute densité ou « HDL », mais aussi dans la
rétroconversion des HDL2 en HDL3 (via l’hydrolyse de différents composants
lipidiques par la lipase hépatique), et vraisemblablement dans la liaison et la
captation de HDL par des récepteurs spécifiques. Enfin, le foie joue un rôle
essentiel dans la récupération des vitamines liposolubles et leur remise en
circulation dans des nouvelles VLDL.
b. Conditions pathologiques
(1) La
stéatose hépatique
Les principaux facteurs causant un engorgement du
foie en triglycérides ou stéatose hépatique sont : un apport accru d’acides
gras (d’origine exogène ou endogène), une stimulation de la lipogenèse
hépatique, et une production réduite d’apoprotéine B-100.
Par définition, un apport exagéré d’acides gras
exogènes résulte de prises alimentaires excessives et est associée à un excès
pondéral.
Une augmentation d’apports d’acides gras endogènes
provient d’une stimulation de leur mobilisation à partir des tissus adipeux
sous l’action de facteurs aussi divers que : les catécholamines (dans les états
de stress), des glucocorticoïdes (dans le syndrome de Cushing ou lors de
traitements à base de stéroïdes), des cytokines cachectisantes comme la TNFa (dans les « wasting diseases »,
d’une résistance à l’action de l’insuline (dans le diabète, mais aussi
communément présente dans la cirrhose), et dans la dénutrition calorique.
La lipogenèse hépatique est stimulée par une
consommation importante d’alcool, un excès d’apports en hydrates de carbone,
l’action de cytokines (comme le TNFa et l’interleukine-1), et
divers agents toxiques (comme le CCl4 et certains médicaments).
La production d’apo B-100 est diminuée dans les
états de résistance de l’action de l’insuline, la dénutrition protéique grave,
et l’insuffisance hépatocellulaire sévère. Dans une proportion importante de
cas aboutissant également à la stéatose, la production totale d’apo B-100 peut
être stimulée, mais dans des proportions insuffisantes que pour compenser
l’importante production hépatique de triglycérides.
(2) Altération
du profil en acides gras essentiels dans les pathologies hépatiques
Dans les insuffisances
hépatocellulaires sévères, on observe fréquemment une carence en acides gras
essentiels à chaînes longues (³ 20 atomes de carbone) des 2
lignées en n-6 et n-3. Cette anomalie reflète une altération du processus
d’élongation-désaturation, et est parfois aggravée par des apports alimentaires
insuffisants ou, plus fréquemment, une nutrition artificielle inadéquate. La
composition en acides gras de nombreuses membranes cellulaires est alors
modifiée, ce qui peut entraîner des perturbations importantes du fonctionnement
membranaire et du métabolisme cellulaire.
(3) Altérations
du profil des lipoprotéines plasmatiques dans les hépatopathies
Les multiples implications du foie à différents
niveaux du métabolisme des lipoprotéines laissent deviner l’hétérogénéité et la
complexité des altérations observées.
La stéatose
est fréquemment accompagnée d’une élévation importante des triglycérides
plasmatiques, présents dans les VLDL à un stade précoce, mais aussi dans les
IDL à un stade plus avancé de l’insuffisance hématocellulaire (ceci étant lié à
une altération de la lipase hépatique).
Dans les cas
aigus d’insuffisance hépatocellulaire, la production d’apo B-100 n’est plus
assurée et l’on peut observer une hypobêtaliprotéinémie (effondrement des VLDL
et des LDL) particulièrement inquiétante chez l’enfant nouveau-né (parce
qu’associée à une grave carence en vitamine E). Dans ces états, on peut
également observer une diminution de production d’apo A-I et (surtout) d’apo
A-II, accompagnant une diminution des HDL plasmatiques (alors que la
consommation chronique d’alcool en quantités modérées est associée à des taux
élevés d’HDL).
Le profil lipoprotéinémique le plus souvent observé
dans l’insuffisance hépatocellulaire
chronique résulte d’une réduction d’activité de la LAT et de la CETP, et
est caractérisé par une diminution des VLDL et des HDL (qui sont pauvres en
cholestérol estérifié et en HDL3). La concentration des LDL est souvent normale,
mais celles-ci sont enrichies en triglycérides et appauvries en cholestérol
estérifié. La dénutrition aggrave souvent la réduction du cholestérol qui
représente un index pronostique intéressant.
La cholestase
est généralement associée à une concentration augmentée de tous les composants
lipidiques du plasma : triglycérides, cholestérol (mais la fraction libre), et
phospholipides, ces deux derniers refluant dans la circulation avec la bile.
L’activité réduite de la lipase hépatique entraîne une augmentation des IDL
(liée à une diminution de leur catabolisme) et une augmentation des HDL2 par
rapport aux HDL3 (liée à une diminution de la rétroconversion). Enfin,
l’association à la cholestase d’une réduction d’activité LCAT aboutit à la
formation d’une lipoprotéine particulière, la lipoprotéine X, qui est trouvée
dans la fraction de densité des LDL, et consiste en un assemblage de
phospholipides et de cholestérol avec des apo C et de l’albumine.
4. Métabolisme des vitamines et micronutriments
Nous ne considérerons ici
que les altérations (carences ou surcharges) en vitamines et oligo-éléments
rencontrés dans le cadre des hépatopathies.
a. Vitamines hydrosolubles
Des carences en vitamines hydrosolubles sont
couramment rencontrées chez les malades présentant une insuffisance
hépatocellulaire, généralement en association avec l’alcoolisme et la
dénutrition. La carence en acide folique
est associée à l’anémie mégalocytaire. La carence en thiamine (vitamine B1) est fréquente chez les alcooliques et est la
cause du syndrome de Wernicke-Korsakoff, de la cardiopathie du béribéri, et
probablement de la polyneuropathie. Des troubles neurologiques, hématologiques
et dermatologiques sont souvent liés à une carence en pyridoxine (vitamine B6) et à une altération de son métabolisme.
Il convient de contrôler ces 3 vitamines chez les
malades alcooliques (en présence ou non d’une insuffisance hépatocellulaire) et
de considérer leur supplémentation.
b. Vitamines liposolubles
Les carences en vitamines liposolubles sont généralement
rencontrées dans les pathologies cholostatiques où elles sont causées par une
malabsorption des graisses. La vitamine
A doit généralement être supplémentée (5.000 - 15.000 Ul/j) chez les
cirrhotiques non alcooliques ; par contre, il convient d’être prudent dans les
pathologies alcooliques où la toxicité de la vitamine A est augmentée (cf. plus
loin).
La carence en vitamine
D est associée à l’ostéoporose et à l’ostéopénie fréquemment présentes chez
les cirrhotiques. Cependant, la supplémentation (100-300 µg/j de 25-OH D3)
n’améliore pas toujours ces altérations osseuses. La carence en vitamines E (a-tocophérol) peut avoir des conséquences dramatiques (altérations neurologiques et
rétiniennes) dans l’hépatopathie sévère du nouveau-né. Bien que le rôle
important de la vitamine E comme agent protecteur contre les dégâts
peroxydatifs soit bien établie, on ne dispose pas actuellement de suffisamment
de données que pour recommander sa supplémentation en routine dans la
cholestase de l’adulte.
La carence en vitamine
K est connue depuis longtemps pour être responsable d’un allongement du
temps de prothrombine, et l’efficacité de la supplémentation est bien étayée.
c. Les oligo-éléments
Une surcharge en cuivre est rencontrée non seulement
dans la maladie de Wilson, mais aussi dans les états cholostatiques. Une
surcharge en fer est rencontrée dans l’hémochromatose génétique, mais aussi
dans la cirrhose alcoolique (cf. plus loin), les maladies hémolytiques, et les
malades en hémodialyse chronique (du moins avant l’utilisation régulière de
l’érythropoïétine). Ces métaux (cuivre et fer) représentent de puissants agents
prooxydants qui peuvent induire ou aggraver les dommages liés à la peroxydation
lipidique.
Une carence en zinc
est fréquemment rencontrée dans les hépatopathies alcooliques.
Rappelons-nous du rôle important du zinc dans les synthèses protéiques, ainsi
que de son influence sur le maintien de l’immunité cellulaire.
B. LA FONCTION BILIAIRE
1. Métabolisme de la bilirubine
a. Origines de la bilirubine
La bilirubine résulte du catabolisme d’hémoprotéines
dans le système réticulo-endothélial.
L’hémoglobine
libérée lors de la destruction physiologique des globules rouges, en est la source
principale (± 80%). Environ 20% de la
bilirubine provient d’hémoprotéines
d’origine hépatique ou rénale (catalase, peroxydase, myoglobine, cytochrome
P450, ...), et moins de 3% de la bilirubine provient d’une érythropoïèse inefficace (érythrocytes détruits dans la moelle
avant leur libération).
b. Métabolisme normal de la bilirubine
La bilirubine
se lie avec une forte affinité à l’albumine. La bilirubine étant liposoluble et liée à
l’albumine ne passe pas dans les urines
même si sa concentration sérique est élevée.
Le foie est l’unique organe qui a la capacité d’éliminer
la bilirubine de la circulation sanguine. La bilirubine est captée par les
cellules parenchymateuses du foie alors que l’albumine reste dans le plasma.
Dans l’hépatocyte, la bilirubine est liée à la
ligandine et est transportée jusqu’au réticulum endoplasmique où elle est
conjuguée à de l’acide glucuronique par l’UDP-glucuronosyl-transférase ce qui
conduit à la transformation séquentielle de la bilirubine en dérivés mono- et
diglucuroconjugués. Cette conjugaison peut être induite par le phénobarbital.
La bilirubine conjuguée est à nouveau transportée
par une ligandine jusqu’au pôle canaliculaire de l’hépatocyte où elle est
excrétée par un transporteur actif. Ce transporteur a une capacité limitée ce
qui entraîne une limite à la captation de l’hémoglobine par l’hépatocyte. 85%
de la bilirubine conjuguée est excrétée dans la bile, le reste reflue dans le
sang.
Dans l’intestin, la bilirubine conjuguée subit un
catabolisme par des enzymes d’origine bactérienne et la majeure partie de la
bilirubine est transformée en urobilinogènes. Le cycle entéro-hépatique de la
bilirubine est mineur. Les urobilinogènes fécaux sont transformés en
stercobiline qui donne aux selles leur couleur caractéristique. Une faible
partie des urobilinogènes est réabsorbée par l’intestin. Une fraction est
éliminée dans les urines.
Les glucuronides de la bilirubine sont solubles dans
l’eau et apparaissent dans les urines lorsque le niveau plasmatique est
augmenté.
c. Pathologie du métabolisme de la bilirubine
(1) Production
augmentée de la bilirubine
En cas d’hémolyse, les quantités de bilirubine
produites dépassent les capacités de captation des hépatocytes et conduit à une
accumulation de bilirubine non conjuguée
dans le sang. Il n’y a pas de bilirubine (urines jaunes) dans les urines.
(2) Déficit
en glucuronyl transférase
Un déficit en glucuronyl transférase conduit à une accumulation dans le sang de bilirubine
non conjuguée. Un tel déficit est observé dans les maladies de Gilbert
(fréquente) et de Crigler-Najjar (rare).
(3) Défaut du
transport canaliculaire de la bilirubine conjuguée
La nature exacte du défaut est mal connue. On
observe une accumulation de bilirubine directe et conjuguée. Ils surviennent
dans la maladie de Dubin Johnson (rare) et la maladie de Rotor (très rare).
(4) Obstruction biliaire
On observe une augmentation du taux de bilirubine portant
principalement sur la fraction conjuguée. De la bilirubine conjuguée est
présente en grande quantité dans l’urine (urines porto).
(5) Altération
des hépatocytes (hépatite, cirrhose)
Les hépatocytes continuent à conjuguer la
bilirubine, mais l’excrétion est diminuée ou abolie. Comme dans le cas
précédent, de la bilirubine s’accumule
principalement sous forme conjuguée avec présence d’une bilirubine
conjuguée.
|
BIOCHIMIE DES ICTERES |
||
|
ETIOLOGIE |
BILIRUBINEMIE |
BILIRUBINURIE |
|
Hémolyse |
Ý non conjuguée |
Absente |
|
ß Glucuronyl-tr. |
Ý non conjuguée |
Absente |
|
Hépatite |
Ýmixte |
Présente |
|
Obstruction |
Ýmixte |
Présente |
2. Métabolisme des acides biliaires
a. Métabolisme normal
Le foie synthétise de l’acide cholique et
désoxycholique à partir du cholestérol. Cette synthèse est contrôlée par la
quantité de sels biliaires réabsorbée au niveau de l’iléon. Elle est
inversement proportionnelle à la quantité d’acides biliaires traversant le foie
par unité de temps. Ces acides sont conjugués (à la taurine et à la glycine) et
éliminés dans la bile sous formes de sels de Na. Après le repas, des quantités
importantes de sels biliaires passent dans l’intestin. 80% de ces sels
biliaires sont déconjugués et réabsorbés au niveau de l’iléon. Ce cycle
entéro-hépatique se produit plusieurs fois par jour. Il explique que bien que
le pool des sels biliaires soit peu important (2 à 3 g), 25 à 30 g de sels
biliaires sont sécrétés dans la bile par 24 heures. Les acides biliaires sont
indispensables à la digestion et à l’absorption des graisses alimentaires. Ils
sont nécessaires à la sécrétion de la bile par l’hépatocyte.
Les sels biliaires non absorbés dans l’iléon sont
déconjugués et déshydroxylés par les bactéries du côlon produisant de l’acide
déoxycholique, de l’acide lithocholique et de l’acide ursodésoxycholique.
b. Pathologie
D’une manière générale, une augmentation du taux de
sels biliaires dans le sérum et les urines apparaît dans les affections hépatiques (obstruction des voies biliaires et
destruction des hépatocytes). Certains sels biliaires, et notamment l’acide
lithocholique, sont hépatotoxiques et provoquent des lésions hépatocellulaires
dans les rétentions biliaires prolongées.
Les sels biliaires sont responsables du prurit observé dans les troubles de la
sécrétion biliaire. Lorsque l’obstruction est incomplète, l’administration
orale d’un chélateur des sels biliaires, la cholestyramine, diminue le prurit.
L’absence de sels biliaires dans l’intestin provoque
une stéatorrhée par défaut de
formation de micelles.
3. La sécrétion biliaire
La formation de la bile (600
ml/24 heures) est la conséquence de la sécrétion active de composés organiques
et inorganiques (bilirubine, acides biliaires, phospholipides, minéraux,
glutathion). Cette sécrétion active est suivie de la filtration osmotique d’eau
et d’électrolytes.
La sécrétion biliaire comprend :
·
Une
sécrétion hépatocytaire et canaliculaire.
·
Une
sécrétion canalaire par l’épithélium des voies biliaires.
a. La sécrétion hépatocytaire et canaliculaire (400 ml/24 heures)
(1) Dépendante des acides biliaires
Elle comprend trois étapes :
·
La
captation (via des transporteurs) par la membrane sinusoïdale de l’hépatocyte.
·
Le
transport intracellulaire jusqu’au pôle canaliculaire de l’hépatocyte.
·
La
sécrétion canaliculaire (via des transporteurs).
(2) Indépendante
des acides biliaires
Elle est liée à la sécrétion active de glutathion et
d’autres composés organiques ou minéraux (bilirubine, phospholipides, …) via également
des transporteurs.
b. La sécrétion canalaire (200 ml/24 heures)
Elle est riche en bicarbonates et influencée par la
sécrétine qui active un transporteur localisé au niveau de la cellule
épithéliale biliaire.
Chacun des
systèmes de transport impliqués dans la sécrétion biliaire peut être altéré par
régulation négative (exemple : médicaments, cytokines, endotoxine, …) ou
des mutations et entraîner des
hyperbilirubinémies ou des cholestases.
III. FOIE ET ALCOOL
A. CONSIDERATIONS GENERALES
L’incidence et la prévalence des hépatopathies
alcooliques varient considérablement entre les pays, mais représente dans nos
pays occidentaux un important problème de santé publique, puisque la majorité
(environ 65%) des pathologies sont causées par l’alcool. Il faut noter
d’importantes différences interindividuelles dans la tolérance à l’alcool, et
notamment en fonction du sexe. On considère généralement que le risque de
développer une pathologie commence avec une consommation moyenne de 40 g/j chez
l’homme et de 20 g/j chez la femme.
Les trois types de lésion hépatique causée par
l’alcool sont : la stéatose, l’hépatite, et la cirrhose. Il existe fréquemment
une association entre les différents types de lésions, et des manifestations
pathologiques extra-hépatiques ne sont pas rares. Ces lésions ne sont
généralement pas causées par l’éthanol lui-même, mais par des composés
résultant de son métabolisme.
B. METABOLISME DE L’ALCOOL
La quantité d’alcool atteignant le foie par unité de
temps est influencée par la vidange gastrique, l’absorption par l’intestin
grêle proximal, et l’extraction hépatique à partir du sans portal. Notons que
l’alcool se distribue dans les compartiments liquidiens de l’organisme (le fait
que ceux-ci sont nettement plus limités chez la femme explique la tolérance
réduite), et n’est pas déposé dans les tissus périphériques.
Le métabolisme de l’alcool est caractérisé par deux
étapes oxydatives qui surviennent essentiellement au niveau du foie. Elles
peuvent être résumées comme suit :
ETHANOL ® ACETALDEHYDE ® ACETATE
1. Production d’acétaldéhyde
La première étape, amenant à la formation du dérivé
toxique l’acétaldéhyde, est catalysée par trois enzymes : l’alcool
déshydrogénase (cytosolique), le système enzymatique microsomal CYP 2EI lié au
cytochrome P450 ou (« microsomal enzymatic oxidative system » ou
MEOS), et la catalase (peroxysomale). Dans des conditions de consommation
d’alcool faible ou modérée, celui-ci est converti en acétaldéhyde, surtout par
l’alcool déshydrogénase (ADH) :
ADH
CH3CH2OH + NAD+ ®® ® ® ® ® ® CH3CHO + NADH + H+
Nous reviendrons plus tard sur l’importance potentielle d’une production importante de NADH dans le cytosol.
L’activité de l’ADH n’est pas stimulée, mais
apparaît au contraire être inhibée par l’alcool. Cependant, les alcooliques
chroniques présentent souvent une tolérance accrue à la consommation de
quantités importantes, liée à une clairance accélérée à l’éthanol à partir du
sang. Ceci résulte de l’induction du MEOS, dont l’activité (et celle d’autres
enzymes microsomales) est fortement stimulée. Le système CYP 2EI réalise la
conversion de l’éthanol en acétaldéhyde comme suit :
MEOS
CH3CH2OH + NADPH + H+ ®®®®®®®® CH3CHO + 2 H2O2 + NADP+
L’action
de la catalase, qui reste quantitativement très limitée, ne sera pas détaillée.
2. Dégradation de l’acétaldéhyde
La dégradation de l’acétaldéhyde en acétate
s’effectue sous l’action de deux acétaldéhydes déshydrogénase (ALDH), l’une
cytosolique (ALDH1), l’autre mitochondriale (ALDH2) nettement prédominante :
ALDH
CH3CHO + NAD+ + H2O ®®®®®®®CH3COO- + NADH + 2H+
Différents facteurs peuvent inhiber le
fonctionnement de l’ALH, et, en réduisant la conversion de l’acétaldéhyde en
acétate, augmenter la concentration locale en acétaldéhyde.
L’alcool lui-même pourrait perturber l’action de
l’ALDH. Des antibiotiques (le céfamandole) possèdent également cette propriété
et leur utilisation doit être assortie de précautions. L’activité ALDH est
diminuée dans les pathologies hépatiques, qu’elles soient d’origine alcoolique
ou non. Enfin, on connaît depuis longtemps l’importance des différences
individuelles et raciales aux effets de l’alcool. Ces différences sont
notamment liées au polymorphisme du gène et de la protéine ALDH2. Une mutation
de ce gène (ALDH2*2), communément rencontrée chez les asiatiques (> 50%),
inhibe l’action de l’enzyme et maintient un taux élevé d’acétaldéhyde. Cette
mutation peut être associée à une activité augmentée de l’ADH, et engendrer
lors de la prise d’alcool, des effets secondaires importants, comme un flush,
une hypotension et une tachycardie (accumulation d’acétaldéhyde). En général,
le type d’isoenzyme ALDH détermine le comportement des individus vis-à-vis de
l’alcool, en induisant une répulsion chez ceux présentant une forme moins
active. Il s’agit d’un mécanisme protecteur car ces individus présentent un
risque accru de dégâts hépatiques.
C. FACTEURS RESPONSABLES DES LESIONS HEPATIQUES
1. L’acétaldéhyde
L’acétaldéhyde est le facteur le plus impliqué dans
le développement des lésions hépatiques. Sa toxicité peut s’exprimer
directement ou indirectement.
De façon
directe, l’acétaldéhyde perturbe le fonctionnement mitochondrial, provoquant ainsi une
altération du métabolisme oxydatif, et à terme la nécrose cellulaire. Notons
que cette toxicité mitochondriale augmente la peroxydation lipidique, induisant
ainsi la formation d’autres aldéhydes, comme la malondialdéhyde. Par ailleurs,
ces perturbations diminuent l’activité ALDH2, et réduisent le catabolisme de
l’acétaldéhyde.
L’acétaldéhyde, comme d’autres aldéhydes, peut se lier à différentes protéines
cellulaires et former des « adducts ». Ces « adducts »
perturbent le fonctionnement des protéines impliquées. Par exemple, la liaison
à la tubuline inhibe la formation de microtubules et altère ainsi la sécrétion
de protéines, tandis que la liaison à l’actine entraîne une augmentation des
dégâts hépatocellulaires. La formation
d’ « adducts » induit la synthèse de collagène au niveau des
cellules de Ito (cf. plus loin). Enfin, ces composés, en modifiant la conformation
des protéines (notamment au niveau des membranes cellulaires), peuvent
représenter des néoantigènes qui entraînent la formation d’anticorps dirigés
contre eux. On assiste alors au développement d’une réaction immunitaire
dirigée contre le foie.
2. Production de NADH dans le cytosol
La production cytosolique de NADH qui y est
normalement présent en quantité très faible, modifie le rapport NAD+/NADH (LI :
~ 1000) et le potentiel rédox. Ceci stimule
les réactions de réduction. La conversion massive de pyruvate en lactate
entraîne une acidose lactique, et inhibe la gluconéogenèse ce qui peut résulter
en une hypoglycémie sévère si les dépôts de glycogène ont été consommés. Par
ailleurs, ces modifications du potentiel rédox induisent une hyperuricémie et
favorisent la formation des triglycérides et le développement de la stéatose.
Elles peuvent également être impliquées dans l’inhibition de synthèse de la
testostérone et la diminution des performances sexuelles (sans diminution de la
libido) dans les états éthyliques.
3. Formation d’acétate
Alors que l’alcool et l’acétaldéhyde restent
essentiellement dans le foie, l’acétate peut être exporté vers les tissus
extra-hépatiques pour y servir de substrat énergétique. Au niveau hépatique,
l’acétate entraîne une dégradation accrue de l’ATP et une déplétion énergétique
des cellules. Ceci résulte en une augmentation de la production de radicaux
libres, notamment via l’induction du métabolisme de l’hypoxanthine par la
xanthine oxydase. Par ailleurs, la formation d’acétate au niveau du foie
stimule la cétogenèse et la lipogenèse, augmentant le risque de stéatose.
4. Peroxydation lipidique
Il est maintenant bien acquis que la peroxydation
lipidique joue un rôle dans les hépatopathies alcooliques, mais son importance
reste encore mal précisée. Ces peroxydation représente un déséquilibre entre la
production de radicaux libres (qui intervient physiologiquement dans de
nombreux processus cellulaires) et les mécanismes antioxydants protecteurs. La
consommation d’alcool augmente la production de radicaux libres et diminue le
potentiel antioxydant.
La production de radicaux libres est stimulée par
l’activation des enzymes microsomaux (cytochrome P450) ainsi que par
l’activation du système xanthine oxydase par la production d’acétate. Par
ailleurs, des métaux comme le fer sont connus pour être de puissants agents
prooxydants. Le contenu en fer du foie est souvent augmenté chez les
alcooliques et l’acétaldéhyde facilite la libération du fer à partir de la
ferritine. Enfin, notons que l’activation des cellules de Küpffer stimule la
production de radicaux libres survenant de façon normale dans le métabolisme de
ces macrophages.
La diminution du potentiel antioxydant est due à une
réduction des mécanismes enzymatiques (qui agissent en convertissant les
radicaux libres), comme la superoxyde dismutase (SOD) et la glutathion
peroxydase, dont le fonctionnement est altéré de façon non spécifique dans
l’insuffisance hépatique, ainsi qu’à la déplétion en agents non enzymatiques (qui
agissent en captant les radicaux libres), comme la vitamine E, la vitamine C,
le sélénium et le glutathion (agissant surtout au niveau intracellulaire).
La peroxydation lipidique provoque des altérations
importantes au niveau des membranes cellulaires qui modifient le métabolisme
normal des hépatocytes, et un clivage (non spécifique) de l’ADN qui se traduit
par une aggravation des dommages hépatiques. Enfin, notons que les aldéhydes
provenant de la peroxydation lipidique inhibent l’activité ALDH dans les mitochondries,
ce qui maintient des concentrations élevées en acétaldéhyde.
D. PERTURBATIONS DU METABOLISME HEPATIQUE
1. Métabolisme énergétique
On observe souvent chez les alcooliques une
augmentation des dépenses énergétiques liée à la production de cytokines
pro-inflammatoires (cf. plus loin). Par ailleurs, le métabolisme oxydatif élevé
de l’alcool (qui consomme beaucoup d’O2) et la déplétion d’énergie induite par
la production d’acétate peuvent entraîner un état d’hypoxie qui va accentuer
sévèrement les effets toxiques de l’alcool.
2. Métabolisme des lipides
Le principal résultat de la consommation d’alcool
est le développement d’une stéatose hépatique. Les principaux facteurs
précipitant ou aggravant celle-ci sont :
·
l’épargne
d’oxydation des acides gras qui sont disponibles pour la ré-estérification.
·
La
modification du potentiel rédox et la prolifération microsomale (avec
activation enzymatique) qui stimulent la formation de triglycérides.
Cette formation accrue de triglycérides entraîne une augmentation de la production de VLDL, et rappelons à cet égard que l’alcoolisme est la cause la plus fréquente d’hypertriglycéridémie. Toutefois, la sécrétion des VLDL est débordée par la production de triglycérides qui s’accumulent ainsi dans les hépatocytes. Par ailleurs, notons les valeurs élevées d’HDL-cholestérol fréquemment trouvées chez les consommateurs d’alcool, avec une prédominance d’HDL2 lorsque leur conversion en HDL3 sous l’action de la lipase hépatique est compromise.
3. Métabolisme des hydrates de carbone
Rappelons simplement le risque d’hypoglycémie lors de la consommation abondante d’alcool, notamment dans le contexte d’un état de jeûne prolongé entraînant la mobilisation prématurée des dépôts de glycogène.
4. Métabolisme des protéines
On observe souvent chez les alcooliques chroniques
une concentration plasmatique abaissée pour l’albumine et les protéines
viscérales. En fait, ceci n’est pas le résultat d’une réduction de la synthèse,
mais bien d’une diminution de la sécrétion causée par les difficultés de
formation des microtubules. Par ailleurs, la dégradation des protéines
intracellulaires par protéolyse dans les lysosomes est également perturbée. La
conjugaison de ces facteurs explique l’accumulation de protéines dans les
hépatocytes, entraînant leur gonflement (« ballooning ») et à terme
leur nécrose.
Notons également une diminution de synthèse (cette
fois) des glycoprotéines et donc des apoprotéines liée à un défaut de la
glycolysation au niveau du système de Golgi.
5. Synthèse de collagène
La consommation chronique d’éthanol entraîne une
fibrose hépatique causée par le dépôt de collagène au niveau des espaces de
Dissé et des hépatocytes environnants. Ce collagène est notamment produit par
les cellules de Ito (cf. anatomie pathologique) dans les régions
périsinusoïdales. Des « adducts » d’acétaldéhyde stimulent la
synthèse de collagène par des cellules de Ito activées. L’activation des
cellules est induite par des dérivés de la peroxydation lipidique, ainsi que
par le « transforming growth factor » (TGF) b1 (cf. plus loin). Par ailleurs, leur
prolifération est stimulée par différentes cytokines comme l’interleukine-1
(IL-1), le TNFa, le « platelet derived
growth factor » (PDGF), l’ « epidermal growth factor »
(EGF).
D’autres facteurs paraissent favoriser la production
de collagène et le développement de la fibrose. Citons notamment l’hypoxie, les
carences nutritionnelles fréquemment associées à l’alcoolisme, ainsi que
l’augmentation du contenu en fer des hépatocytes.
6. Production de cytokines
La production de nombreuses cytokines est associée à
la présence de lésions hépatiques chez les alcooliques, mais le fait de savoir
si leur libération est causée directement par l’alcool ou résulte d’un effet
non spécifique lié au développement d’une pathologie n’est pas bien précisé.
La concentration de cytokines inflammatoires (IL-1,
TNFa, IL-6, IL-8) est nettement augmentée dans le
foie et dans le plasma lors d’hépatites alcooliques aiguës, et ce, en relation
directe avec la sévérité de l’atteinte. Lors de la régression du processus
pathologique, la concentration d’IL-6 et d’IL-8 se normalise rapidement alors
que celle d’IL-1 et de TNFa se maintient à des taux
élevés de façon prolongée. Cette stimulation de la production de cytokines
inflammatoires peut être causée par des radicaux libres, l’hypoxie
intralobulaire et par l’arrivée au foie d’endotoxines traversant une barrière
intestinale à la perméabilité augmentée.
Les « transforming growth factor » (TGF)
sont impliqués dans la fibrinogenèse à partir des cellules de Ito stimulées.
Leur production semble être plus en rapport avec le développement de la maladie
qu’avec un effet direct de l’alcool. Ils possèdent aussi des propriétés
immuno-suppressives.
Nous avons déjà mentionné l’effet du PDGF sur la
prolifération des cellules de Ito. Son rôle in
vivo est suggéré par la présence de substances « PDGF-like » dans
le liquide d’ascite.
7. Régénération hépatique
Divers facteurs participent
à altérer la régénération du foie chez les malades alcooliques, comme on
l’observe en clinique après des résections hépatiques. Il semble bien que les
modifications de composition (et de structure) des membranes plasmatiques des
hépatocytes (résultant notamment de la peroxydation lipidique et de la
formation d’ « adducts » sur les protéines membranaires) jouent
un rôle déterminant en perturbant le fonctionnement normal des ligands et des
récepteurs, ainsi que l’expression des protéines G. Le résultat est une réponse
inadéquate aux facteurs hépatotrophiques, comme les mitogènes et co-mitogènes
ainsi que les protooncogènes agissant habituellement sur les cellules
hépatiques.
8. Conséquences de l’activité accrue du système microsomal
En dehors de la peroxydation lipidique déjà
discutée, l’induction d’activité du système microsomal augmente la production
et hépato-toxicité de l’acétaldéhyde. Par ailleurs, cette induction enzymatique
augmente nettement la sensibilité du foie aux solvants organiques et à certains
médicaments (par exemple paracétamol) mais aussi à des agents carcinogènes.
Elle augmente également la toxicité de la vitamine A et la dégradation de
plusieurs micronutriments.
CHAPITRE II :
METHODES D’EXPLORATION
EN PATHOLOGIE HEPATIQUE
I. APPROCHE BIOCHIMIQUE DE LA PATHOLOGIE HEPATIQUE
A. LES TESTS HEPATIQUES CONVENTIONNELS
1. Indice de captation, conjugaison, et excrétion hépatique
a. Bilirubine et dérivés de la bilirubine
La bilirubine réagit avec certains sels de diazonium pour former des asodérivés colorés.
La bilirubine conjuguée réagit directement avec le colorant, tandis que la bilirubine non conjuguée doit être séparée de l’albumine pour réagir. Cette séparation peut être obtenue par de la caféine.
|
INTERPRETATION D’UNE
HYPERBILIRUBINEMIE. |
||
|
Ý BILIRUBINE NON CONJUGUEE Enzymes hépatiques normaux |
Ý BILIRUBINE CONJUGUEE ET NON CONJUGUEE Enzymes hépatiques
anormaux |
|
|
Ý réticulocytes |
Réticulocytes N. |
CYTOLYSE ou CHOLOSTASE |
|
HEMOLYSE |
MALADIE DE GILBERT |
|
b. Acides biliaires
Ils peuvent être estimés quantitativement au moyen
d’un dosage radio-immunologique.
Les principales indications de leur détermination
sont :
·
L’estimation
de la sévérité d’une affection hépatique parenchymateuse chronique (cirrhose,
hépatite chronique).
·
L’estimation
du pronostic (survie) chez les cirrhotiques.
2. Indice de lésions hépatocellulaires (Cytolyse)
a. Aspartate-(AST) et alanine aminotransferase
(ALT)
L’AST (ou ASAT), également appelée transminase
oxalo-acétique glutamique (GOT ou SGOT), car elle catalyse la réaction :
aspartate + a-oxoglutarate ® oxalo-acétate + glutamate. Elle est
retrouvée dans le cytoplasme et les mitochondries de nombreux tissus comme le
foie, le myocarde, les muscles squelettiques et les globules rouges.
L’ALT (ou ALAT), également appelée transminase
glutamique pyruvate (GPT ou SGPT), car elle catalyse la réaction : alanine a-oxoglutarate ® pyruvate + glutamate. Elle
est confinée au cytoplasme et les concentrations les plus élevées sont
retrouvées dans le foie.
|
ENZYMES INTRA CELLULAIRES |
||||
|
Concentration
dans les organes (par ordre croissant) |
||||
|
ALT |
AST |
¡ GT |
LDH |
CPK |
|
Foie Rein Cœur Muscle |
Cœur Foie Muscle Rein Pancréas |
Foie Rein Pancréas Voies biliaires |
Muscle Foie Rein Myocarde Ganglions. GR. Rate Cœur Pancréas |
Muscle (MM) Cœur (MB) Cerveau (BB) |
Lorsque les
tissus sont lésés, avec une augmentation de la perméabilité membranaire et de
la nécrose cellulaire, ils libèrent ces enzymes dans le sang.
|
ENZYMES LIBERES DANS LE SANG |
||||
|
Pathologies concernées |
||||
|
ALT |
AST |
¡ GT |
LDH |
CPK |
|
Cytolyse
hépat. Infarctus
myoc. Hypothyroïdie |
Infarctus Myoc. Cytolyse
hépat. Pancréatites Nécrose muscles Hypothyroïdie |
Cytolyse
hépat. Cholostase Pancréatites Aff. rénales |
Infarctus Myoc. Nécrose muscles Cytolyse
hépat. Aff. hématol Pancréatites Aff. rénales |
Infarctus Myoc. Nécrose muscles |
|
INTERPRETATION D’UNE AUGMENTATION DES TRANSAMINASES |
|||||
|
|
ALT |
AST |
PAL |
¡ GT |
CPK |
|
CYTOLYSE |
|
|
N |
|
N |
|
CHOLOSTASE |
|
|
|
|
N |
|
INFARCTUS MYOCARDE |
|
|
N |
N |
|
En pathologie hépatique, le rapport ASAT/ALAT est
< sauf dans les cas suivants :
·
Hépatite
alcoolique.
·
Evolution
d’une hépatite chronique vers une cirrhose.
Atteinte hépatique aiguë sévère.
b. Lactate déshydrogénase (LDH)
Les LDH sont retrouvées dans le
cytoplasme de nombreux tissus. Leur spécificité est donc faible, mais la
détermination des isoenzymes permet d’augmenter cette spécificité. En effet,
l’isoenzyme LDH5 est augmentée de façon préférentielle dans les affections
hépatiques. Néanmoins, l’existence d’autres enzymes facilement dosables dans le
sang, plus sensibles et plus spécifiques a limité l’intérêt du dosage de la LDH
ou même de la LDH5 dans les affections hépatiques.
3. Indice de lésions canalaires ou enzymes de cholestase
a. Phosphatases alcalines (PAL)
Il s’agit d’un ensemble d’enzymes dont
l’électrophorèse permet de séparer des isoenzymes de différentes origines.
Le taux de PAL est au-dessus des valeurs de
référence de l’adulte durant la période
néonatale, mais aussi durant la phase
de croissance osseuse c’est-à-dire l’adolescence, en cas de maladie de
Paget, d’ostéomalacie ou de métastases osseuses. Les PAL augmentent également à
partir du troisième mois de grossesse
(PAL d’origine placentaire) pour atteindre des valeurs doubles des valeurs
usuelles chez l’adulte, en fin de grossesse.
L’activité des
PAL totales sériques augmente dans de nombreuses maladies hépato-biliaires, mais
les taux les plus élevés sont retrouvés en cas d’obstacles à l’écoulement de la
bile. Dans
ces cas, l’élévation des PAL n’est pas liée à un défaut d’excrétion hépatique
de l’enzyme, mais bien à une augmentation de synthèse notamment sous l’action
des acides biliaires. De même ces acides biliaires solubilisent les PAL
membranaires qui atteignent le plasma par régurgitation paracellulaire ou par
endocytose transcellulaire.
b. g
Glutamyl transférase
La gGT
est une glycoprotéine qui catalyse le transfert du groupe gGlutamyl
à partir des peptides gGlutamyl, comme le glutathion, sur d’autres peptides,
acides aminés ou l’eau.
Elle se retrouve dans toutes les membranes qui ont
une activité de sécrétion ou d’absorption élevée (activité maximale dans le
rein, le foie et le pancréas).
Chez le sujet normal, l’activité sérique de gGT est principalement d’origine hépatique.
Les valeurs de référence varient selon la technique utilisée, mais sont plus
élevées pour le sexe masculin et sont très vite élevée en période néonatale et
chez l’enfant jusqu’à l’âge d’un an. Il faut savoir qu’environ 5% de la
population présente une activité sérique en gGT supérieure à la normale
sans affection sous-jacente retrouvée.
Le taux de gGT augmente dans toutes les maladies hépatiques, c’est donc un test
très sensible mais ayant une faible valeur diagnostique.
|
ETIOLOGIES D’UNE ELEVATION DES gGT
sériques |
||
|
Avec des PAL |
Avec des ALAT et ASAT |
Isolée |
|
CHOLOSTASE Intra ou extra-hépatique |
CYTOLYSE Aiguë ou chronique |
Sujet normal Médicaments inducteurs
enzymatiques (Barbit.) Alcoolisme Hyperthyroïdie Parasitoses |
Son utilisation dans la prise en charge de sujets
alcooliques est largement répandue. En effet, probablement induite par
l’alcool, la gGT voit son activité sérique
augmenter dans cette population. Néanmoins, il existe une mauvaise corrélation
entre la consommation d’alcool et l’activité sérique de la gGT.
c. Leucine aminopeptidase (LAP) et
5’nucleotidase (5’nase)
Ces deux enzymes sont présentes dans tous les tissus humains, mais seules certaines affections hépatiques, et la grossesse pour la LAP alors d’origine placentaire, provoquent une augmentation de leur activité dans le sang. Le dosage de la g-glutamyl transférase a largement supplanté leur dosage.
Leur utilisation principale reste
la confirmation qu’une élévation des PAL est bien d’origine hépatique.
4. Indice de synthèse hépatique
Le foie synthétise de très nombreuses protéines circulantes. Leur concentration ne dépend pas seulement de ce niveau de synthèse, mais aussi de nombreux facteurs extra-hépatiques : demi-vie des protéines individuelles, leur catabolisme, leur volume de distribution.
a. Protéines totales
Les altérations du taux des protéines totales sont
peu spécifiques, elles peuvent apparaître dans nombre d’affections non
hépatiques. De ce fait, l’estimation du taux des protéines totales n’a que peu
de valeur en hépatologie.
b. Electrophorèse des protéines
On observe des modifications de l’électrophorèse
dans certaines situations (par exemple élévation de la bande des gGlobulines dans l’hépatite chronique active),
mais cette analyse a un faible intérêt
diagnostic en hépatologie.
c. Albumine et g
globulines sériques
Une diminution du taux d’albumine avec élévation du
taux de gamma-globulines est observée dans les insuffisances hépatiques
chroniques (cirrhoses).
d. Préalbumine
C’est une protéine qui se lie à des iodothyronines
et une molécule de retinol binding protein (RBP). En raison de sa courte
demi-vie (1,9 j), ses modifications sont plus précoces que celles de l’albumine
(demi-vie : ± 20 j). Il faut toutefois se
souvenir que la préalbumine comme l’albumine est une protéine
« négative » de la phase inflammatoire : elle voit ses concentrations
diminuer lors d’une phase inflammatoire, ce qui en limite son utilisation dans
de nombreuses situations. C’est également un marqueur de l’état nutritionnel.
e. Immunoglobulines
On observe une augmentation des IgA dans les
cirrhoses, des IgG dans les hépatites chroniques et des IgM dans la cirrhose
biliaire primitive.
f. Temps de prothrombine (PT) et dosage du
facteur V
Le PT, ou temps de QUICK (anciennement PTT), permet
d’évaluer la voie intrinsèque de coagulation (facteurs I, II, V, VII, IX et X),
c’est-à-dire d’évaluer des facteurs de coagulations synthétisés par le foie. Sa
valeur est exprimée en pourcentage de la normale. SI la fonction hépatiques est déficiente, le temps de prothrombine sera
allongé. Des déterminations successives peuvent être utilisées pour suivre
la progression de la maladie ou pour évaluer le risque hémorragique d’un
patient.
Le PT est allongé ans les troubles de la résorption de la vitamine K (cholostase,
stéatorrhées). L’administration parentérale de vitamine K corrige le PT en cas
de malabsorption de la vitamine K mais pas en cas d’insuffisance de synthèse
hépatique. Une manière plus sophistiquée, mais non utilisées en routine
clinique, de diagnostiquer la cause d’un allongement du PT consiste à doser le
facteur V dont la production ne dépend pas de la vitamine K.
g. Cholinestérase
C’est une enzyme qui hydrolyse une variété d’esters
de la choline. Les concentrations
diminuent dans les affections hépatiques, et surtout la cirrhose, en étant
le reflet d’une diminution de synthèse hépatique.
En cas de polymorphisme
génétique, des taux bas peuvent être observés et ceci est détectable par un
trouble de l’inhibition de l’activité cholinestérasique par la dibucaïne ou le
fluorure.
h. Cholestérol sérique
Le taux de cholestérol est, en
général, augmenté en cas de cholostase et diminué en cas d’insuffisance
hépatique, mais il existe de nombreuses causes non hépatiques d’hyper ou
d’hypocholestérolémie ce qui limite l’intérêt de la détermination du taux de
cholestérol en pathologie hépatique.
B. LES TESTS QUANTITATIFS EXOGENES
1. Principes
Les tests hépatiques « conventionnel » apportent des renseignements sur l’intégrité du foie et sur la formation de la bile, mais peu donnent une information sur la fonction hépatique. Diverses substances exogènes peuvent elles être utilisées pour explorer le fonctionnement global du foie. Ces substances évaluent soit la capacité de synthèse hépatique (administration d’un substrat et suivi de la formation du produit synthétisé), soit la capacité de clairance hépatique. Par analogie à la clairance rénale, la clairance hépatique reflète la quantité du composé extrait de la circulation générale par le foie par unité de temps, relative à la concentration du composé dans le sang atteignant le foie. La quantité extraite peut être calculé : Cl hépatique = flux sanguin x extraction hépatique.
En pratique, tous les tests exogènes reflètent la perfusion hépatique, la capacité fonctionnelle et les échanges hépatocyte-sang. Mais certains tests dépendent principalement de la perfusion hépatique (clairance du galactose à faible dose, du vert d’indocyanine), tandis que d’autres dépendent initialement de la capacité fonctionnelle hépatique (clairance du galactose à forte dose, de la caféine, déméthylation de l’aminopyrine).
Mais en pratique, ces deux types
de tests donnent des résultats moins différents que la théorie le laissait
prévoir. Il existe une hétérogénéité des fonctions métaboliques hépatiques de
telle sorte que la clairance d’un seul composé n’évalue que partiellement ce
métabolisme, il existe une réserve fonctionnelle hépatique énorme, et enfin il
existe aussi une intertrication des altérations fonctionnelles et des shunts.
2. Valeur clinique des tests exogènes
a. Indice
diagnostique
Il existe une grande variabilité individuelle des résultats chez les sujets normaux, mais pas intraindividuelle, ce qui permettrait plutôt d’interpréter des résultats sériés chez un même individu. Ces tests ne permettent pas, bien entendu, de porter un diagnostic étiologique, ils évaluent la réserve fonctionnelle hépatique.
b. Indice de
sévérité et pronostic de la cirrhose
On pourrait croire que ces tests
puissent apporter une information pronostique indépendante d’autres facteurs
comme le score de Child-Pugh, mais ceci est controversé (voir chapitre sur la
cirrhose). Une étude longitudinale, c’est-à-dire des prélèvements sériés dans
le temps pour un même patient, semble être une approche d’avenir.
II. L’IMAGERIE
A. L’ECHOTOMOGRAPHIE
L’utilisation d’ultrasons
permet d’explorer la morphologie du foie, des vaisseaux hépatiques et des voies
biliaires de manière non invasive et pour un prix de revient inférieur aux
autres procédés. Cette méthode permet de déceler des lésions focales, liquides
et solides et des anomalies d’échostructure du parenchyme hépatique. Des
quantités peu importantes d’ascite sont décelées avant l’apparition de signes
cliniques. Il est en outre possible d’évaluer, avec l’aide du Doppler, le
calibre des vaisseaux ainsi que la présence et la direction d’un flux et
éventuellement d’évaluer son débit et sa vitesse.
B. LA TOMODENSITOMETRIE
Ce procédé complète d’échotomographie. Il fournit
des renseignements sur le volume et la densité du foie. Après injection rapide
de produit de contraste et acquisition de coupes précoces, il peut détecter des
tumeurs hypervascularisées et permet l’exploration des vaisseaux hépatiques.
C. LA RESONANCE MAGNETIQUE
Ce procédé récent peut être utile lorsque des
méthodes classiques ne permettent pas un diagnostic certain, notamment en cas
de lésion focale. Il permet aussi l’exploration des vaisseaux hépatiques.
Dans les surcharges en fer, le foie apparaît foncé si on le compare au muscle.
Des techniques récentes permettent la projection
spatiale de l’arbre biliaire et pancréatique sans injection de produit de
contraste pour autant qu’il y ait dans les structures bilio-pancréatiques,
assez de liquide non circulant.
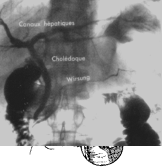
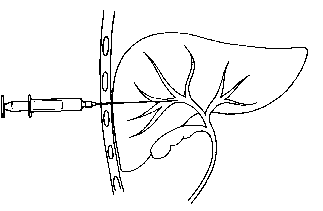
D. LA CHOLANGIO-PANCREATOGRAPHIE ENDOSCOPIQUE ET LA CHOLANGIOGRAPHIE TRANSPARIETALE
La localisation précise d’une cause de cholostase
nécessite souvent l’opacification radiologique de l’arbre biliaire et des
canaux pancréatiques. Les méthodes utilisées seront décrites en détail dans le
chapitre consacré aux voies biliaires.
E. L’ANGIOGRAPHIE
L’injection sélective de produit de contraste dans le tronc coeliaque ou même dans l’artère hépatique permet dans un premier temps de visualiser les artères, dans un deuxième temps les capillaires (phase parenchymateuse) et dans un troisième temps, les veines. Les « masses » hépatiques sont visualisées et leur irrigation éventuelle précisée.
L’angiographie permet la caractérisation de lésions vasculaires hépatiques, la mise en évidence de fistules artério-biliaires ou artério-portales et complète souvent la mise au point d’une hypertension portale, particulièrement avant chirurgie.
Une cavographie complétée par une
opacification des veines sus hépatiques peut être réalisée.
III. LA LAPARO(COELIO)SCOPIE
La laparo(coelio)scopie permet d’explorer, sous
anesthésie locale, la cavité péritonéale, la morphologie du foie (taille,
anomalies de couleur et de surface) et de détecter les signes d’hypertension
portale.
Des biopsies peuvent être prélevées sous contrôle
visuel avec possibilité d’électrocoagulation en cas d’hémorragie.
IV. LA BIOPSIE HEPATIQUE ET LA CYTOPONCTION
L’absence de corrélation parfaite
entre les résultats des tests biologiques et la morphologie du foie rend
nécessaire la réalisation de biopsies hépatiques non seulement pour préciser le
diagnostic d’une affection hépatique et sa sévérité mais également pour en
surveiller l’évolution. Ces biopsies
peuvent être réalisées soit à l’aveugle, soit au cour d’une laparoscopie, soit
par voie trans-jugulaire. Ces examens se font en ambulatoire après contrôle
de la coagulation (PT > à 50%, APTT dans les normes, Plaquettes >
50.000/mm³) sauf pour la biopsie trans-jugulaire. En cas d’ascite, une vidange
préalable à l’examen est indispensable. Après biopsie à l’aveugle ou par
laparoscopie, il est indispensable de surveiller le rythme cardiaque et la
tension artérielle afin de déceler la survenue éventuelle d’une hémorragie.
La biopsie
aveugle est surtout indiquée dans les lésions diffuses du foie ou dans le
cadre d’une mise au point d’une altération des tests hépatiques. Une
échographie doit être réalisée au préalable.
La biopsie
transveineuse est réalisée par abord jugulaire droit. Elle peut être
réalisée en cas de troubles de l’hémostase et permet la mesure simultanée du
gradient sus hépatique.
La cytoponction
sous contrôle échographique ou tomodensitométrie permet de faire des
prélèvements à l’aiguille fine destinés à une étude cytologique. Ce procédé est
surtout utile au diagnostic des lésions focales.
CHAPITRE III :
MANIFESTATIONS CLINIQUES :
LES GRANDS SYNDROMES
Les manifestations cliniques des maladies hépatiques
comportent 3 grands syndromes :
·
L’insuffisance
hépatique.
·
La
cholostase.
·
L’hypertension
portale.
Une seule maladie peut comporter l’association de
plusieurs syndromes (insuffisance hépatique et hypertension portale dans la
cirrhose, cholostase et insuffisance hépatique dans la cholostase chronique).
I. L’INSUFFISANCE HEPATIQUE
L’insuffisance hépatique est l’ensemble des signes
liés à la diminution, voire à l’arrêt, des fonctions de synthèse hépatocytaire.
A. L’INSUFFISANCE HEPATIQUE CHRONIQUE
Les principales étiologies de l’insuffisance hépatique chronique sont
reprises dans le Tableau suivant.
|
ETIOLOGIES DE L’INSUFFISANCE HEPATIQUE CHRONIQUE |
|
|
Etiologies |
Exemples |
|
Pathologie parenchymateuse Pathologie vasculaire Pathologie cholestatique sévère |
Cirrhoses Hépatite chronique sévère Syndrome de Budd Chiari Maladie veino-occlusive Cirrhoses biliaires |
L’insuffisance hépatique chronique peut n’entraîner
aucune manifestation clinique. Son diagnostic repose alors sur des anomalies
biologiques. La symptomatologie est disparate et dépend en grande partie de
l’intensité et de la durée de l’insuffisance hépatique.
|
MANIFESTATIONS CLINIQUES DE L’INSUFFISANCE HEPATIQUE CHRONIQUE SEVERE |
|
|
|
Exemples |
|
1.
Asthénie et malnutrition 2.
Ictère 3.
Troubles neuropsychiques 4. Ascite 5. Troubles rénaux 6.
Manifestations cutanées 7. Signes
endocriniens 8.
Troubles hématologiques 9.
Manifestations cardio-vasculaires et
pulmonaires 10. Susceptibilité aux infections |
Encéphalopathie hépatique Syndrome hépatorénal Angiomes stellaires Erythème palmaire Hypogonadisme, gynécomastie Aménorrhée, stérilité Epistaxis, purpura, hémorragies débit cardiaque, ¯ T.A. Syndrome hépato-pulmonaire Péritonite bactérienne spontanée |
1. Asthénie et malnutrition
Elles sont liées à la diminution des apports, à
l’anorexie, à la malabsorption, aux troubles du métabolisme des nutriments et à
l’augmentation des besoins nutritifs (chez l’alcoolique).
2. Ictère
L’insuffisance hépatique peut être responsable d’un
ictère de degré variable.
Une certaine corrélation existe entre l’intensité de
l’ictère et la gravité de l’insuffisance parenchymateuse. Cette donnée n’est
cependant pas absolue. Dans la cirrhose par exemple, l’ictère peut être absent.
Sa présence est néanmoins un signe de mauvais pronostic.
3. Encéphalopathie
a. Définition
L’encéphalopathie hépatique représente l’ensemble
des manifestations neuropsychiques secondaires à l’arrivée au cerveau de
substances neurotoxiques produites par l’intestin et non catabolisées par un
foie malade ou passant dans la circulation générale en raison d’anastomoses
porto-caves.
b. Pathogénie
Ammoniac : l’ammoniac, produit dans le côlon à partir des protéines sous l’influence des enzymes bactériennes, n’étant plus métabolisé en urée par le foie malade ou shunté, arrive dans le cerveau et est détoxifié dans les astrocytes en glutamine ce qui induit un oedème cérébral.
Récepteur GABA/Benzodiazépine
Il existe au niveau cérébral un récepteur GABA (acide gamma-amino-butyrique) benzodiazépine qui, lorsqu’il est occupé, entraîne un influx de chlore et une inhibition de la neurotransmission.
L’activation peut être induite par le GABA lui-même (produit dans l’intestin) ou des benzodiazépines endo- ou exogènes.
Manganèse : une accumulation de
manganèse dans le globus pallidus pourrait être responsable de signes
extrapyramidaux, s’observant souvent en cas d’encéphalopathie hépatique.
c. Manifestations cliniques
Une encéphalopathie peut apparaître sur un terrain
d’hépatopathie chronique au stade de cirrhose ou dans l’évolution d’une
insuffisance hépatocellulaire aiguë grave.
Il existe deux formes d’encéphalopathie :
Aiguë
: Elle peut être spontanée (de mauvais pronostic) ou déclenchée par un facteur
précipitant : infection, hémorragie, troubles électrolytiques, excès de
protéines, constipation, benzodiazépines.
Chronique : Cette forme est en rapport avec les shunts porto-systémiques
naturels ou créés chirurgicalement. On peut observer des fluctuations
neuro-psychiques selon l’apport de protéines dans l’alimentation.
Si on classe la symptomatologie en fonction du degré
de gravité des manifestations, on décrit classiquement 4 stades :
stade 1 : apathie, irritabilité ou au contraire jovialité inadaptée,
diminution del’attention et impossibilité de réaliser des calculs simples
(addition, soustraction).
stade 2 : désorientation dans le temps, comportement inadéquat, modification
de la personnalité, confusion.
stade 3 : somnolence, état stuporeux, désorientation sévère.
stade 4 : coma.
La recherche des signes et tests suivants facilite
la confirmation de l’origine hépatique des symptômes précités :
·
L’astérixis est démontré en provoquant
une extension du poignet, l’avant-bras étant tenu immobile. Les doigts sont
écartés les uns des autres. En cas d’encéphalopathie, on observe, de façon plus
ou moins fréquente, des mouvements de flexion-extension des articulations
métacarpo-phalangiennes.
·
Le
fétor hépatique ou haleine de foie
cru, correspond à l’exhalation d’une substance probablement d’origine
intestinale.
·
Les
tests de piste : ces test plus
complexes et plus sensibles que le test de l’étoile consistent à faire relier
par un trait soit 25 cercles numérotés de 1 à 25, soit un chiffre à une lettre
(1-A, 2-B, 3-C) de 1 à 13 (A à M) et de calculer le temps nécessaire pour réaliser
ce travail correctement.
d. Diagnostic paraclinique
L’association d’un syndrome neurologique non
systématisé et de signes en faveur d’une hépatopathie suggère fortement le
diagnostic d’encéphalopathie hépatique.
Parmi les examens complémentaires on retiendra :
·
L’électroencéphalogramme
: cet examen met en évidence une diminution plus ou moins importante de la
fréquence du rythme alpha normal (8 à 13 cycles par seconde) remplacé par des
ondes lentes de haut voltage. Ce type de tracé peut être observé dans diverses
altérations de type métabolique (urémie, acidose respiratoire) et être observé
précocement avant toute manifestation clinique ou biologique.
·
Le
dosage du NH3 artériel : observée
dans des conditions techniques précises, le taux d’ammoniaque artériel augmente
dans les maladies hépatiques chroniques ou aiguës sévères, mais il existe une
mauvaise corrélation entre le taux d’ammoniaque et le degré de
l’encéphalopathie.
·
Le
bilan biologique hépatique. Il
mettra en évidence des signes d’insuffisance hépatique (diminution de
l’albuminémie, du temps de prothrombine, etc...).
e. Diagnostic différentiel
Le coma peut ressembler à tous les autres types de coma métabolique. Il
convient également d’exclure d’autres causes de coma, comme la méningite
suppurée, l’hémorragie méningée et
l’hématome sous-dural survenant à la suite d’un traumatisme.
Des symptômes neuropsychiques peuvent également
survenir chez les alcooliques présentant soit un delirium tremens, soit une encéphalopathie
de Wernicke. En cas de delirium tremens, on observera en général une
hyperactivité motrice continue, des hallucinations et de l’insomnie.
L’encéphalopathie de Wernicke s’accompagne de paralysie des nerfs oculomoteurs,
de strabisme et de troubles de l’équilibre.
L’utilisation de diurétiques chez les cirrhotiques
peut entraîner de l’hyponatrémie.
Cette altération peut engendrer des symptômes neuropsychiques en l’absence de
coma hépatique. Il s’agit le plus souvent d’apathie, de somnolence, de
céphalées et d’hypotension. Outre une hyponatrémie, une augmentation du taux de
l’urée est fréquente.
f. Traitement
·
L’apport protéique sera diminué, mais il
ne doit pas tomber en dessous de 60 g de protéines par jour. En général, les
protéines végétales sont mieux tolérées, et des suppléments d’acides aminés
branchés per os sont préconisés si l’on veut augmenter l’apport protéique sans
aggraver l’encéphalopathie.
·
Les antibiotiques non
résorbables
La néomycine et le métronidazole
(Flagyl â) sont efficaces. Ils ne
peuvent être prescrits que durant de courtes périodes à cause de leur toxicité.
Ils détruisent la flore bactérienne productrice d’ammoniaque.
·
Les disaccharides non
digestibles
Le lactulose, (Bifitéral â), a progressivement
remplacé la néomycine responsable d’effets secondaires relativement fréquents.
Le lactulose est métabolisé dans l’intestin. Les lactates formés favorisent
l’excrétion fécale de l’ammoniaque. Le lactitol
(Portolac â) est un dissacharide de
deuxième génération se présentant sous forme de poudre en sachets. La dose doit
être ajustée en vue de provoquer deux selles semi-liquides par jour. Lactulose
et Lactitol peuvent être administrés en lavements en cas d’hémorragie ou de
manque de collaboration du malade.
4. Ascite
L’ascite peut survenir brutalement à la suite d’une
dégradation rapide de la fonction parenchymateuse comme par exemple au décours
d’une hémorragie, d’une infection, d’une débauche éthylique. Elle peut
apparaître progressivement après une période plus ou moins prolongée de
ballonnement.
Cf. § IV L’ASCITE.
5. Syndrome hépato-rénal
Le syndrome hépato-rénal est une insuffisance rénale
fonctionnelle progressive par vasoconstriction avec oligoanurie survenant chez
des patients cirrhotiques décompensés. Il peut être favorisé par
l’administration non contrôlée de diurétiques. On n’observe aucune lésion
anatomique des reins et leur transplantation à des sujets exempts d’affection
hépatique est suivie d’une restauration de la fonction rénale.
Le syndrome hépato-rénal est caractérisé par une
oligurie et une élévation de la créatininémie (> à 1,5 mg/dl) avec absence
d’amélioration suite à l’arrêt des diurétiques et à la perfusion d’1 l 500
d’expanseur plasmatique et en dehors d’un contexte d’état de shock, de
déshydratation, d’infection ou de prise de médicaments néphrotoxiques. Le
sédiment urinaire est en général banal. En fonction de ces données, il est
généralement aisé de différencier ce syndrome d’autres causes d’insuffisance
rénale comme par exemple, la nécrose tubulaire aiguë secondaire à une
hémorragie ou à une infection ou à une insuffisance rénale prérénale (par
déshydratation).
Le traitement est, en général, décevant en dehors de la transplantation hépatique et de la combinaison d’expanseurs plasmatiques et de glypressine (dérivé de la vasopressine).
6. Manifestations cutanées
a. Angiomes stellaires
Il s’agit de lésions
sous-cutanées, punctiformes, formées d’une artériole centrale d’où partent de
petits vaisseaux ressemblant à des pattes d’araignées.
b. Erythème palmaire
Ce signe consiste en une rougeur pommelée, localisée
sur les éminences thénar et hypothénar ; il n’est pas spécifique : il peut
être observé dans l’arthrite rhumatoïde et dans les maladies fébriles
chroniques. Il est dû à une vasodilatation des capillaires sous-cutanés.
7. Signes endocriniens
Chez l’homme, on observe une féminisation
caractérisée par une gynécomastie,
une perte de phanères (pilosité à caractère féminin), associée à une
augmentation du taux d’oestrone. En outre, un hypogonadisme est responsable
d’une atrophie testiculaire. Celle-ci est associée à une diminution du taux de
testostérone. Une impuissance est également fréquente. Ces manifestations sont
principalement observées dans la cirrhose.
Chez la femme, la diminution d’oestradiol et de
progestérone provoque la perte des caractères
sexuels secondaires comme la graisse pelvienne et mammaire ainsi que la suppression des ovulations. On note en
outre une diminution de sécrétion des gonadotrophines.
8. Troubles hématologiques
Les manifestations des altérations de l’hémostase
dans l’insuffisance parenchymateuse hépatique sont diverses : épistaxis, purpura, hémorragies digestives,
fibrinolyse post-opératoire.
De multiples facteurs interviennent :
· La réduction de la synthèse des facteurs de coagulation : II, V, VII, IX et X. La synthèse du fibrinogène est également diminuée mais est plus résistante que celle des autres facteurs.
· Une coagulation intravasculaire disséminée : la nécrose hépatique peut provoquer une activation de l’hémostase, en même temps qu’une diminution de la clairance des facteurs activés. Une consommation locale du fibrinogène, des plaquettes et d’autres facteurs se manifeste. La fibrinolyse locale provoque la formation de produits de dégradation du fibrinogène. Le résultat final de ces altérations consiste en hémorragies. Ce type de coagulopathie est caractérisé par une diminution du fibrinogène plasmatique, une hypoplaquettose, une diminution des facteurs de coagulation.
· Une fibrinolyse primaire : ce mécanisme plus rare peut intervenir dans des situations aiguës (trauma, interventions). L’activation de la fibrinolyse peut résulter d’une diminution de la clairance hépatique d’un activateur du plasminogène libéré par exemple au cours d’une intervention.
· Une cholostase peut aggraver la situation en réduisant l’absorption intestinale de la vitamine K dont dépend la synthèse des facteurs II, VII, IX et X.
·
L’insuffisance hépatique chronique peut, par ailleurs,
entraîner des modifications des globules rouges, blancs et des plaquettes.
Avant le traitement d’une hémorragie, il convient
d’en préciser le(s) mécanisme(s) responsable(s). Une carence en vitamine K sera
corrigée par l’administration parentérale à la dose de 10 mg/jour. En général,
ce traitement sera peu efficace à cause de l’insuffisance parenchymateuse
hépatique. Les mécanismes responsables d’une coagulation intravasculaire
disséminée doivent être recherchés et traités: infection, choc, déshydratation.
Les transfusions de plaquettes sont efficaces pour une courte période. On a
également recommandé l’utilisation de plasma frais congelé ou de concentré de
complexe prothrombinique (facteurs II, VII, IX et X) avant un acte invasif.
9. Altérations cardio-vasculaires et pulmonaires
a. Cardio-vasculaires
L’insuffisance hépatique chronique provoque un état
d’hypercinésie cardiaque, conséquence de la diminution des résistances
vasculaires périphériques. Ce syndrome se manifeste par de la tachycardie, une augmentation du débit cardiaque et une baisse de la pression artérielle
diastolique.
b. Pulmonaires
Dans environ 30% des cas de cirrhose, on observe une
hypoxémie artérielle en l’absence de toute affection cardio-pulmonaire
organique. Elle se manifeste par de la dyspnée
parfois aggravée en position assise (platypnée)
et de la cyanose. C’est le syndrome
hépato-pulmonaire.
Dans les cas graves, cette vasodilatation pulmonaire
excessive provoque un shunt artério-veineux et une altération des mécanismes de
ventilation-perfusion.
10. Sensibilité aux infections
En présence d’une pyrexie chez un cirrhotique ou
d’une cirrhose décompensée, il convient d’éliminer une cause infectieuse
bactérienne.
B. INSUFFISANCE HEPATIQUE AIGUE SEVERE
L’insuffisance hépatique aiguë sévère représente
l’ensemble des manifestations cliniques associées à une dysfonction
hépato-cellulaire brutale et sévère (PT < 50%), survenant dans un foie
préalablement normal.
En cas de survie, la structure et les fonctions
hépatiques redeviennent normales.
Les causes de l’insuffisance hépatique sévère sont
reprises dans le tableau suivant :
|
ETIOLOGIE DE L’INSUFFISANCE HEPATIQUE AIGUE SEVERE |
|
|
|
Exemples |
|
Virus (± 70%). Médicaments et toxiques. Vasculaires. Diverses. |
A, B, B delta, C, Herpès…. Paracétamol, INH, acide valproïque. Halothane et dérivés. CCl4. Amanite phalloïde. Syndrome de Budd-Chiari. Maladie veino-occlusive. Hypoxie. Métastases, leucémies, lymphomes. Stéatose aiguë gravidique. Maladie de Wilson. Syndrome de Reye. |
1. Manifestations cliniques
Elles sont similaires à celles observées en cas
d’insuffisance hépatique chroniques et comprennent surtout :
a. Ictère
Une certaine corrélation
existe entre le degré d’ictère et la gravité de l’insuffisance hépatique.
b. Encéphalopathie
Une encéphalopathie est une manifestation reflétant
toujours la gravité de l’insuffisance hépatique. Elle peut se développer
endéans les 2 semaines (hépatite fulminante) ou au-delà des 2 semaines
(hépatite subfulminante) suivant le début de l’ictère. Contrairement à ce qui
se passe dans l’encéphalopathie de l’insuffisance chronique (où les shunts
porto-cave jouent un rôle important), le facteur : oedème cérébral joue ici un
rôle prépondérant entraînant une hypertension intracrânienne.
Les signes cliniques seront ceux de
l’encéphalopathie (grade I à IV) et de l’oedème cérébral : convulsion, rigidité
musculaire, mouvements de décérébration et de décortication, anomalies des
réflexes pupillaires.
c. Coagulopathie
On note une réduction des
facteurs de coagulation synthétisés par le foie (PTT, facteur V) ainsi qu’une
éventuelle coagulation intravasculaire disséminée (æ fibrinogène, æ plaquettes, ä produits de dégradation du
fibrinogène).
d. Autres manifestations
On peut aussi observer une hypercinésie circulatoire
systémique, un syndrome hépatopulmonaire, un syndrome hépatorénal et une
susceptibilité excessive aux infections ainsi qu’une hypoglycémie (liée à la
diminution des réserves de glycogène).
2. Traitement
Tout patient atteint d’une insuffisance hépatique
sévère doit être hospitalisé en unité de
soins intensifs où l’on préviendra et traitera les complications de
l’insuffisance hépatique aiguë et un contact doit être pris avec un centre de
transplantation.
Une attention particulière doit être apportée à la
détection et au traitement d’un oedème
cérébral. En cas d’hypertension intracrânienne, on propose l’administration
de mannitol à 20% (1g/kg de poids) pour autant qu’il n’y ait pas d’insuffisance
rénale. Si une perfusion de 1 à 2 l de dextrose I.V. est généralement
suffisante, la survenue d’une hypoglycémie
peut rendre nécessaire l’utilisation de concentrations plus élevées (20 à 50%).
Parallèlement, des doses suffisantes
d’électrolytes, et particulièrement de potassium, seront administrées. Les
indications habituelles de dialyse
sont applicables aux insuffisances rénales associées à cette situation.
Une coagulopathie
peut nécessiter l’administration de plasma frais congelé, de plaquettes et de
vitamine K (seulement s’il y a un syndrome hémorragique ou avant un acte
invasif). Le maintien d’un pH gastrique au-dessus de 5 (par les anti H2) évite
les épisodes d’hémorragies gastriques.
Il convient de maintenir
une Pa CO² à 4-4,5 Kpa par une ventilation appropriée.
On surveillera particulièrement la survenue
d’épisodes septiques en vue d’utiliser l’antibiothérapie
indiquée. Une désinfection intestinale est recommandée.
L’acétyl-cystéine IV est indiquée dans le
traitement de l’insuffisance hépatique sévère induite par le paracétamol à
hautes doses et pour combattre l’hypoxie cellulaire car elle apporte la
cystéine nécessaire à la synthèse du glutathion.
Dans la plupart des cas, seule la transplantation hépatique peut sauver
le malade. La sélection des patients prend en compte des données cliniques
(degré d’encéphalopathie) et biologiques (importance de l’élévation de la
bilirubinémie et de la chute du PTT).
II. LA CHOLOSTASE
On désigne sous de nom de
cholostase l’ensemble des manifestations liées à la diminution ou à l’arrêt de
la sécrétion biliaire. Comme les altérations sécrétoires isolées de la
bilirubine ne répondent pas à la définition de la cholostase, elles seront
traitées séparément.
A. ANATOMOPATHOLOGIE
1. Cholostase obstructive (extra ou intra-hépatique)
L’obstruction des voies biliaires intra et
extra-hépatiques aboutit à une stase de la bile qui s’accumule en périphérique
de l’acinus de Rappaport, à la fois dans les hépatocytes et dans les
canalicules biliaires situés entre les hépatocytes. L’accumulation dans les
canalicules distend ces derniers, formant en coupe de petits amas arrondis de bile appelés thrombus biliaires, tandis que
l’accumulation intracellulaire augmente la pression osmotique du cytoplasme
produisant des cellules ballonnées (4 à 10 fois plus grands que les hépatocytes
normaux) que l’on qualifie de cellules en dégénérescence spumeuse. Au niveau de
l’espace porte apparaît, après quelques jours, un oedème important qui
s’accompagne d’un infiltrat marginal de polynucléaires et d’une prolifération de petits canaux biliaires de
néoformation. L’ensemble se fibrose progressivement ; le pontage des
espaces portes agrandis peut réaliser une dissection nodulaire du foie
(cirrhose biliaire secondaire). Les lésions hépatocellulaires ne s’observent
qu’après une cholostase prolongée. La levée de la cholostase permet d’observer
la régression des lésions hépatiques potentiellement réversibles.
2. Cholostase non obstructive
Dans la cholostase dite fonctionnelle, la cause se
situe au niveau des hépatocytes eux-mêmes ou des canalicules biliaires. Le plus
souvent, cette cholostase n’est qu’une des manifestations d’un trouble profond
du parenchyme hépatique (hépatite virale, hépatite éthylique, ...).
B. CLASSIFICATION ET ETIOLOGIE
1. La cholostase obstructive extra-hépatique
La cholostase extra-hépatique est secondaire à une
obstruction des voies biliaires extra-hépatiques. Les causes les plus
fréquentes de cholostase extra-hépatique sont la lithiase, les tumeurs du
pancréas et la pancréatite chronique.
2. La cholostase obstructive intra-hépatique
Elle est due à une obstruction des voies biliaires
intrahépatiques (cf. tableau).
3. La cholostase non obstructive
Elle est due à un arrêt de la formation de la bile
au niveau des hépatocytes. Les causes les plus fréquentes sont les hépatites et
la cirrhose biliaire primitive (cf. tableau).
C. MANIFESTATIONS CLINIQUES
A l’inverse des atteintes parenchymateuses, l’état
général est ben conservé au début de la maladie. Il évolue en fonction de la
cause de la cholostase et s’altère rapidement si l’origine est cancéreuse.
ETIOLOGIE DE LA CHOLOSTASE |
|
A. CHOLOSTASE
EXTRA-HEPATIQUE 1. Lithiase cholédocienne. 2. Tumeurs. *ampullome vatérien,
cancer des voies biliaires *cancer du pancréas 3. Pancréatite chronique. 4. Lésions inflammatoires des voies biliaires. *sténoses post-opératoires *cholangite sclérosante *atrésie des voies biliaires (enfant.) 5.
Parasitoses : ascaris, distomatose, échinococcose. B. CHOLOSTASE
INTRAHEPATIQUE a) avec
obstruction mécanique. 1. Métastase hépatiques 2. Cancer primitif du
foie... 3. Cholangiocarcinome 4. Cholangite sclérosante 5. Maladie polykystique du foie b) sans
obstruction mécanique 1. Hépatites
(médicamenteuses, virales, toxiques, bactériennes) 2. Cirrhose biliaire
primitive 3. Cholostase post-opératoire aiguë bénigne 4. Grossesse 5. Cholostase idiopathique récurrente bénigne 6. Granulomatoses (tuberculose, sarcoïdose) 7. Alimentation parentérale 8. Hémopathies (lymphomes) |
1. Ictère
L’ictère est une manifestation essentielle mais
inconstante de la cholostase. Intense et progressif dans les obstacles complets
(cancers extra-hépatiques, pancréatites chroniques), il peut être fluctuant
(lithiase cholédocienne) à peine ébauché, ou totalement absent (certains
cholostases intrahépatiques).
L’ictère cholostatique s’accompagne d’une
surcoloration des urines et, éventuellement, d’une décoloration des selles.
2. Prurit
Le prurit et les lésions dermatologiques de grattage
qui l’accompagnent sont avec l’ictère les principales manifestations cliniques
de la cholostase. Le prurit peut précéder l’ictère de plusieurs mois ou années
(cirrhose biliaire primitive), de quelques semaines (cholostases néoplasiques
intrahépatiques) ou apparaître en même temps que l’ictère (hépatite, cholostase
extra-hépatique).
On considère que la rétention des sels biliaires est
responsable de ce symptôme. En faveur de cette hypothèse, l’utilisation des
chélateurs des sels biliaires, la cholestyramine, diminue le prurit.
3. Malabsorption
Une stéatorrhée par déficience de sels biliaires
dans la lumière intestinale se développe proportionnellement à l’intensité de
l’ictère lorsque celui-ci se prolonge. Il en résulte une carence en vitamines
liposolubles et en calcium. La carence en vitamine D et en calcium est
responsable d’une ostéoporose et d’une ostéomalacie. Des douleurs osseuses, des
fractures et des tassements vertébraux en constituent l’expression clinique. La
carence en vitamine K est le plus souvent subclinique.
D. LOCALISATION DE LA CHOLOSTASE
Les manifestations
biologiques de la cholostase sont
indépendantes du siège de l’obstruction. Il faut donc se tourner vers
d’autres arguments pour le diagnostic.
(1) arguments cliniques
·
Vésicule
distendue : présence d’un obstacle biliaire sous le cystique.
·
Triade
température, douleur, ictère : angiocholite.
·
Ictère
cholostatique progressif : néoplasie.
(2) arguments d’imagerie
·
Echographie.
·
Cholangiographie
RMN.
·
Cholangiographie
rétrograde per-endoscopique.
·
Cholangiographie
trans-hépatique.
·
Histologie
hépatique.
E. TRAITEMENT
1. Traitement de la cause de cholostase
Les lésions biliaires et pancréatique seront
traitées soit chirurgicalement, soit endoscopiquement (cf. pathologie biliaire
et pancréas).
2. Traitement des conséquences de cholostase
a. Diététique et carences vitaminiques
L’apport calorique doit être suffisant. Des
triglycérides à chaîne moyenne (absorbés sans devoir passer par la phase
micellaire) seront préconisés en cas de stéatorrhée.
Les vitamines liposolubles (A, D, K) seront
administrées, souvent à titre de prévention d’une carence par malabsorption.
b. Carence calcique
Des suppléments de calcium sous forme de lait
écrémé, de fromage blanc et de carbonate de calcium (1,5 g/jour) sont indiqués.
c. Prurit
La cholestyramine (Questran â : 6 à 10 g par jour au début, la dose d’entretien étant 3 g), est le médicament le plus efficace. Les effets secondaires consistent en nausées et en stéatorrhée. La cholestyramine peut interférer avec l’absorption intestinale de vitamine K et de certains médicaments comme la digitoxine.
L’acide ursodéoxycholique (Ursochol â, Ursofalk â 10 à 20 mg/jour)
reconstitue un pool de sels biliaires moins toxiques. Outre son effet sur le
prurit, il améliore la biologie et l’histologie.
III. L’HYPERTENSION PORTALE
Il existe une hypertension portale lorsque le gradient de pression entre la veine porte
et la veine cave est supérieur à 6 mm de Hg. Les symptômes de
l’hypertension portale résultent essentiellement du développement d’une
circulation collatérale court-circuitant le foie et formée de veines dilatées.
A. ETIOLOGIE
L’hypertension portale est presque toujours due à un
obstacle situé sur la circulation porto-hépatique. Cet obstacle peut siéger en
dessous du foie sur la veine porte (bloc infrahépatique), dans le foie (bloc
hépatique) ou au-delà du foie (bloc suprahépatique). Suivant la position du
bloc par rapport au sinusoïde, on parlera de bloc pré-sinusoïdal, de bloc sinusoïdal
ou de bloc post-sinusoïdal.
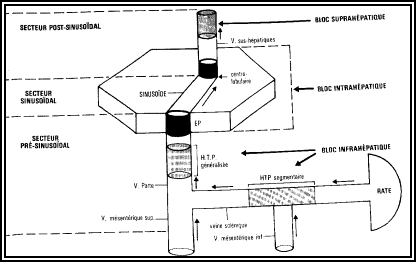
Figure 5.
|
ETIOLOGIES DE L’HYPERTENSION PORTALE |
||
|
Type de bloc |
Exemple |
Caractéristiques |
|
1. Blocs infra-hépatiques |
||
|
Thrombose portale Invasion tumorale porte Compression extrinsèque |
Syndromes
myélo-prolifératifs Déficits en antithrombine
III, protéines C, protéine S Pylephlébite Hépatome Pancréatopathies |
Foie = normal Gradient* = normal |
|
2. Blocs hépatiques |
||
|
Pré-sinusoïdal Sinusoïdal Post-sinusoïdal |
Schistosomiase Cirrhose biliaire
primitive Fibrose congénitale Cirrhose alcoolique Intoxication à la vit. A Hépatite alcoolique Maladie veino-occlusive |
Foie = anormal Gradient* = normal Foie = anormal Gradient* ä Foie = anormal Gradient* ä |
|
3. Blocs supra-hépatiques |
||
|
Budd-Chiari primitif Budd-Chiari secondaire |
Syndrome
myélo-prolifératif Hémoglobinurie
paroxystique nocturne Hépatome, cancer rénal |
Foie = anormal Gradient* = normal Veinographie anormale |
* Déterminé par la différence entre la pression sus
hépatique bloquée et libre.
1. Les blocs infrahépatiques
Ils peuvent résulter :
·
De
thromboses portales et spléniques
survenant au cours de syndrome myélo-prolifératifs ou comme complication d’une
infection dans le territoire porte. Chez l’enfant, la phlébite de la veine
ombilicale peut se compliquer de thrombose porte.
·
D’une
compression de la veine porte ou de
la veine splénique par une tumeur, un kyste, etc. Dans cette catégorie, nous
pouvons ranger les hypertensions portales secondaires aux pancréatites et aux
tumeurs pancréatiques. Ces lésions peuvent provoquer une sténose de la veine
splénique. L’hypertention portale est,
dans ce cas, segmentaire et limitée au territoire splénique.
Dans certains blocs infra-hépatiques, une
circulation collatérale porto-portale s’établit. Elle est formée d’un réseau de
veines dilatées entourant la veine porte (cavernome portal). Les conséquences
métaboliques de ce type d’hypertension portale sont moins importantes.
2. Les blocs hépatiques
Les cirrhoses, quelle que soit leur étiologie, sont
dans notre pays la cause la plus fréquente d’hypertension portale. Le bloc est
surtout sinusoïdal et partiellement post-sinusoïdal.
3. Les blocs supra-hépatiques
Le syndrome de Budd-Chiari
(thrombose ou compression des veines sus-hépatiques) résulte le plus souvent
d’un syndrome myélo-prolifératif, plus rarement d’une compression par une
tumeur hépatique ou rénale.
4. L’excès de flux sanguin portal
Se voit dans les cas de fistule artério-veineuse
entre l’artère hépatique et la veine porte ou entre l’artère et la veine
splénique.
B. MANIFESTATIONS CLINIQUES
1. La circulation collatérale
L’obstacle à la libre circulation du sang portal
provoque la dérivation de ce sang vers les veines formant des communications
entre les systèmes cave et porte. Ces veines se dilatent pour former des
réseaux variqueux.
a. Dérivation supérieure
La conséquence clinique la plus importante de
l’hypertension portale est le développement de varices gastriques (cardio-tubérositaires) et oesophagiennes.
Celles-ci peuvent, si le gradient dépasse 12 mm de Hg, se rompre, provoquant
des hémorragies digestives parfois massives. Outre leurs conséquences
hémodynamiques, ces hémorragies digestives aggravent l’insuffisance hépatique
et peuvent provoquer l’apparition d’un coma hépatique. La mortalité des
hémorragies par rupture de varices oesophagiennes chez les cirrhotiques se
situe entre 40 et 70%.
b. Dérivation antérieure
Le développement inconstant d’une circulation
collatérale par le ligament rond vers les veines ombilicales provoque
l’apparition d’un réseau veineux superficiel péri-ombilicial désigné sous le
nom de « tête de méduse »
ou de syndrome de Cruveilhier et Baumgartner (s’il existe un souffle associé).
Le courant y est centrifuge à partir de l’ombilic alors que dans l’obstruction
de la veine cave inférieure, la circulation collatérale part latéralement de
l’aine vers le gril costal avec un flux sanguin de bas en haut.
c. Dérivation inférieure
Elle draine le sang de la veine mésentérique
inférieure vers le rectum (varices)
et les veines hémorroïdales (hémorroïdes).
d. Dérivation postérieure
Elle provoque une
augmentation de débit à travers les anastomoses pléno-rénales préexistantes et
contribuent à la formation de varices
gastriques (cardio-tubérositaires).
2. L’ascite
Cf. ascite.
3. L’encéphalopathie
L’association de shunts intra ou extra-hépatiques et
d’une insuffisance hépatocellulaire aboutit à une encéphalopathie.
4. La splénomégalie
Elle est secondaire à la congestion et à
l’hyperplasie du tissu réticulo-endothélial. Souvent asymptomatique, elle peut
être responsable d’hypersplénisme qui se caractérise par une anémie, une
leucopénie et une hypoplaquettose. L’hypersplénisme
serait lié à des phénomène de stase, de séquestration et de phagocytose. Une
splénomégalie peut être observée dans les affections hépatiques en dehors de
toute hypertension portale (hépatite virale, mononucléose infectieuse).
5. La gastropathie hypertensive
Cette gastropathie est une manifestation
fonctionnelle spécifique de l’hypertension portale. L’aspect endoscopique est
celui d’un réseau réticulé blanchâtre accompagné de plaques érythémateuses et
de pétéchies. Cette gastropathie est responsable de 10 à 20% des saignements
digestifs compliquant l’hypertension portale.
C. DIAGNOSTIC
Plusieurs méthodes peuvent être utilisées pour
confirmer l’existence d’une hypertension portale et pour déterminer son
origine.
1. Endoscopie
La fibroscopie
permet la visualisation des varices oesophagiennes et gastriques ainsi que les
gastropathies d’hypertension portale. La laparoscopie
permet de visualiser des dilatations des veines épiploïques. En outre, elle est
utile pour confirmer ou infirmer l’existence d’une lésion hépatique.
2. Imagerie
L’échotomographie
éventuellement couplée au Doppler est utilisée pour son caractère non invasif.
Elle permet de visualiser la circulation collatérale, d’évaluer le diamètre de
la veine porte et la vitesse du flux, de déceler la présence d’ascite ou d’une
splénomégalie. La RMN est une
méthode prometteuse pour l’évaluation des vaisseaux.
3. Mesure de la pression portale
La méthode habituellement utilisée pour mesurer la
pression portale consiste en une détermination de la pression régnant dans les
sinusoïdes hépatiques. A cet effet, un cathéter est introduit par voie
périphérique et conduit sélectivement dans une veinule sus-hépatique. S’il
obstrue complètement cette veinule, il permet une mesure de la pression
sinusoïdale (pression sus-hépatique
bloquée). La pression post-sinusoïdale ou pression sus-hépatique libre
s’obtient en retirant progressivement le cathéter. La différence entre ces deux
pressions constitue le gradient sus-hépatique (N£ 6 mm de Hg). Celui-ci est
élevé en cas de cirrhose éthylique (bloc sinusoïdal et post-sinusoïdal) mais
pas dans les hypertensions portales pré-sinusoïdales (bloc infra-hépatique ou
schistosomiases par exemple).
4. Visualisation radiologique de la circulation portale
Elle confirme l’hypertension portale (visualisation
de la circulation collatérale et, éventuellement, flux portal centrifuge),
permet la mise en évidence de la localisation du bloc portal et démontrer
l’existence éventuelle de lésions (thromboses, compressions) des vaisseaux
portes. Tous ces renseignements sont indispensables pour poser les indications
des thérapeutiques chirurgicales. Cette visualisation se fait par artériographie sélective mésentérique et/ou
coeliaque (le temps de retour veineux visualise la circulation porte).
D. TRAITEMENT
Le traitement de l’hypertension portale est, en
général, limité à la prévention ou au contrôle de l’hémorragie par rupture de
varices oesophagiennes. Plus rarement, on sera amené à traiter l’ascite ou
l’hypersplénisme.
1. Prévention primaire
Les malades ayant des varices oesophagiennes à haut
risque de rupture mais qui n’ont jamais saigné seront traités par des
médicaments : bêta-bloquants non sélectifs (propranolol) qui diminuent le débit
cardiaque et le débit collatéral portosystémique. Ils diminuent le risque
d’hémorragie et ont un effet bénéfique sur la survie. En cas de contre-indication, on proposera des ligatures
endoscopiques.
2. Traitement de l’hémorragie aiguë
Le but est de contrôler l’hémorragie, prévenir la
récidive et réduire la mortalité.
a) Réanimation
La correction
de l’hypovolémie par des colloïdes
est indispensable. Des transfusions de sang sont parfois nécessaires. Leur
volume sera aussi limité que possible (il faut obtenir un hématocrite optimum
de 25-30%) afin d’éviter d’induire une récidive hémorragique.
La résorption massive d’ammoniaque résultant de la
dégradation par la flore intestinale du sang présent dans le tube digestif,
joue un rôle important dans le coma hépatique. Pour prévenir cette résorption,
il faut évacuer le sang au moyen de lavements et administrer du lactulose ou du lactilol (cf. traitement de l’encéphalopathie).
Une infection bactérienne étant fréquente chez le
cirrhotique qui saigne, une antibiothérapie empirique est préconisée.
b) Drogues vasoactives
La somatostatine et la glypressine (un analogue de
la vasopressine) sont des vasoconstricteurs splanchniques. Administrées par voie veineuse dès
l’admission et pendant quelques jours, couplées au traitement endoscopique,
elles contribuent à l’arrêt de l’hémorragie.
c) Traitement endoscopique
On utilise la sclérose (produit sclérosant
injecté dans et autour de la varice qui saigne), les ligatures (varice
aspirée dans un cylindre et mise en place d’une ligature élastique à sa base)
ou les colles biologiques (surtout indiquées en cas de varices
gastriques).

Figure
6 La sonde de Linton est
utile en cas d’exsanguination. Le ballon unique, en
forme de poire, est gonflé avec 600 ml d’air et la sonde est mise en
traction (1 kg). La durée de la compression ne doit pas dépasser 12 heures. En
cas d’hémorragies réfractaires au traitement endoscopique on peut envisager
la réalisation d’un shunt
intra-hépatique par voie jugulaire (pour les patients Child B ou C
c’est-à-dire ayant une mauvaise fonction hépatique)) ou un traitement chirurgical (si Child A
c’est-à-dire ayant une bonne fonction hépatique).
En
cas d’exanguination, on utilisera la sonde de Linton (fig. 6).
3. Prévention de la récidive hémorragique
Celle-ci étant fréquente, on recommande après
contrôle de l’hémorragie initiale, d’entreprendre un programme d’éradication
des varices par de séances répétées de ligatures endoscopiques couplées à un
traitement médicamenteux par bêta-bloquants non sélectifs (propranolol).
4. Hémorragie réfractaire ou récidivante malgré les drogues vasoactives et le traitement endoscopique
a) Le shunt porto-cave par voie transjugulaire
en cas de cirrhose Child B ou C
|
|
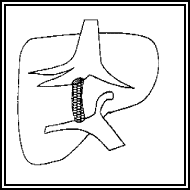
Figure 7
Une
prothèse est placée par voie transjugulaire entre une veine sus-hépatique et la
branche droite de la veine porte, ce qui permet de décomprimer le système
porte.
b) Traitement chirurgical : en cas de
cirrhose Child A
Deux types d’interventions peuvent être réalisés :
les anastomoses porto-caves et les transplantations hépatiques.
|
|
Figure 8
Parmi les anastomoses porto-caves, l’anastomose
porto-cave termino-latérale est la plus anciennement utilisée.
Techniquement assez aisée à réaliser, elle assure
une bonne décompression portale mais se complique à long terme
d’encéphalopathies (30%) parfois invalidantes (17%).
L’anastomose
spléno-rénale proximale (implantation après splénectomie du bout proximal
de la veine splénique dans la veine rénale).
|
|
Figure 9
Elle permet de réaliser une dérivation porto-cave
partielle. Elle se complique moins souvent d’encéphalopathie mais est moins
efficace dans la prévention des hémorragies.
L’anastomose mésentérico-cave
(interposition d’une prothèse en Téflon d’un calibre de 8 mm entre la veine
mésentérique et la veine cave).
|
|
Figure 10
Elle assure une bonne protection contre les
hémorragies au prix d’une fréquence raisonnable d’encéphalopathie. Elle a
l’avantage, par rapport à l’anastomose porto-cave latéro terminale, de ne pas
rendre plus complexe une transplantation hépatique ultérieure.
Lorsque l’obstacle siège sur la veine splénique, la splénectomie supprime totalement
l’hypertension portale. Il en va de même de la cure chirurgicale des anévrismes
artério-veineux du système portal.
La transplantation hépatique est le
meilleur traitement définitif puisqu’elle assure un traitement à la fois de
l’insuffisance hépatique et de ses complications.
IV. L’ASCITE
A. PATHOGENIE
L’ascite ne se développe généralement que lorsque
deux conditions sont réunies :
·
Une
hypertension portale.
·
Une
maladie hépatique.
Dans l’hypertension portale isolée, comme par
exemple chez certains malades atteints de blocs infra-hépatiques, l’ascite est
peu fréquente et modérée. Une rétention hydrosodée isolée provoque un anasarque
et non une simple ascite : c’est l’hypertension portale qui localise la
rétention hydrosodée au péritoine.
L’augmentation de la résistance portale dans
l’hypertension portale est due à des raisons mécaniques (réduction de l’espace
vasculaire par fibrose, capillarisation des sinusoïdes ou hypertrophie des
hépatocytes) et à des raisons fonctionnelles (vasoconstriction liée à une
prolifération des myoblastes, hypersensibilité à la sérotonine ou augmentation
des substances vasoconstrictrices telles que l’endothéline 1). L’augmentation
de la pression portale n’est pas seulement liée à une augmentation des
résistances mais est aussi déterminée par une augmentation du débit efférent
portal splanchnique qui vise à compenser les effets d’une vasodilatation dans
le but de maintenir à tout prix la vascularisation hépatique. Cette
vasodilatation est induite par une quantité excessive de substances
vasodilatatrices (glucagon, substance P, oxyde nitrique, ...). Cette
vasodilatation s’accompagne de phénomènes compensateurs : une augmentation du
débit splanchnique et périphérique (circulation hyperdynamique) et une
stimulation du système rénine-angiotensine-aldostérone avec rétention
hydrosodée.
Dans la cirrhose compensée, cette rétention hydro-sodée est inhibée dès que le
remplissage vasculaire est restauré. Dans la cirrhose décompensée, l’état de
remplissage suffisant tendant à compenser la vasodilatation ne peut plus être
atteint, l’hyperactivation de la rétention se poursuit avec décompensation
ascitique et ses répercussions rénales (insuffisance rénale et syndrome
hépato-rénal).
B. MANIFESTATIONS CLINIQUES
On note une augmentation du volume de l’abdomen
associée à une prise de poids ainsi qu’une matité des flancs encadrant la
sonorité préombilicale.
Une infection du liquide d’ascite peut survenir
spontanément (environ 10%) ou être secondaire à des ponctions : il s’agit
de la péritonite bactérienne
spontanée. Le plus souvent, il s’agit de cirrhoses décompensées. Les
organismes responsables sont généralement des bacilles gram négatifs. La
symptomatologie consiste en douleurs abdominales, pyrexie et sensibilité de
l’abdomen à la palpation.
C. DIAGNOSTIC
L’échographie permet de déceler la présence de 200
ml d’ascite et est donc l’examen de choix pour le diagnostic.
|
CAUSES D’ASCITE |
|
|
1. TRANSSUDAT (PROTEINES < 30 G/L) 2. EXSUDAT (PROTEINES ³ 30 G/L) 3. CHYLEUSE (LACTESCENTE) |
CIRRHOSE INSUFFISANCE CARDIAQUE
GLOBALE NEOPLASIE TUBERCULOSE INSUFFISSANCE
VENTRICULAIRE DROITE PERICARDITE CONSTRICTIVE PANCREATITE SYNDROME DE BUDD-CHIARI COMPRESSION OU OBSTRUCTION DES VOIES LYMPHATIQUES |
En vue de préciser la cause de l’ascite, outre les données cliniques, l’examen du liquide d’ascite obtenu par ponction est fondamental. Il comporte les éléments suivants :
·
aspect : un liquide trouble plaide
en faveur d’une infection bactérienne du liquide. Une ascite sanglante est en
général associée à une affection cancéreuse. Une ascite chyleuse correspond à
une obstruction des lymphatiques et est en général secondaire à une tumeur
maligne ou à un lymphome.
·
examen microscopique : dans une infection
bactérienne, le taux de leucocytes est supérieur à 500/mm³, et le taux de
polynucléaires est supérieur à 250/mm³. Dans la tuberculose, on observe une
prédominance lymphocytaire.
·
examen bactériologique : outre les cultures
habituelles (10 ml d’ascite dans un flacon à hémoculture), qui mettent en
évidence un germe gram négatif dans 2/3 des cas et un pneumocoque dans 1/3 des
cas, on recherche la présence de B.K.
·
cytologie : la recherche de cellules
malignes exige l’utilisation de colorations spéciales et éventuellement de
l’immunocytochime compte tenu des difficultés à différencier les cellules
néoplasiques des cellules endothéliales.
·
examen chimique : la mesure du taux de
protéines permet de séparer les liquides d’ascite en transsudats (moins de 30 g
de protéines/l) et exsudats (plus de 30 g de protéines par litre). Une
imprécision existe entre 20 et 30 g/l.
La mesure du gradient de concentration albumine sérique/albumine ascitique est en corrélation avec la pression portale et peut aider au diagnostic différentiel de la cause d’ascite. Il est inférieur à 11 g/l lorsque la pression portale est normale et supérieur à 11 g/l dans les ascites dues à une hypertension portale.
Le dosage de la LDH peut
éventuellement être utile dans les cas litigieux. En effet, lorsque son taux
dans le liquide d’ascite excède de façon importante le taux sanguin, on peut
suspecter soit une affection cancéreuse, soit une tuberculose.
D. TRAITEMENT
1. Traitement médical
Le principe général du
traitement est de réduire les apports en
sodium et d’en favoriser l’excrétion
urinaire.
Si la rétention hydro-saline est modérée, le traitement peut être commencé ambulatoirement. En cas d’ échec (± 80% des cas) le malade doit être hospitalisé. Le traitement peut être considéré satisfaisant si une perte de poids de 1kg/j est obtenue lorsqu’on observe de l’ascite et des oedèmes et de 500 g par jour lorsqu’il n’y a que de l’ascite.
Le régime
comportera un maximum de 1 à 2 g de sel (40 à 80 mEq de Na) par jour, en
évitant d’excéder la quantité excrétée dans l’urine. Cette réduction des
apports en sel peut être obtenue en excluant les aliments salés et en
supprimant toute adjonction de sel aux aliments.
Une restriction
hydrique (maximum 1 l à 1,5 l/jour) n’est indiquée que chez les patients
présentant une hyponatrémie et chez ceux dont le poids augmente malgré le
traitement médical.
Le repos au
lit peut favoriser la mobilisation des liquides en augmentant la perfusion
rénale.
|
TRAITEMENT DE L’ASCITE |
|
1. Régime
sans sel (40 à 80 mEq/j) et repos au lit. 2. Diurétiques Spironolactone (Aldactone â) (100-400 mg/j) Furosémide (Lasix â) (40-120 mg/j) 3. Ascite réfractaire Paracentèse + Albumine (8
g/l) Shunt intra-hépatique par
voie trans-jugulaire |
Des diurétiques doivent être fréquemment associés aux mesures
diététiques. La présence d’une insuffisance rénale représente une
contre-indication à l’utilisation des diurétiques. Les électrolytes sanguins
doivent être contrôlés 2 fois par semaine.
Plusieurs groupes de diurétiques
sont disponibles. Les diurétiques agissant sur le tube distal, inhibent la
réabsorption du sodium et la sécrétion de K+ et de H+. Ils réalisent donc une
épargne de potassium. Parmi ceux-ci, l’antagoniste
de l’aldostérone (Aldactone â) est le plus souvent utilisé. Les diurétiques agissant sur la branche ascendante de Henle inhibent le
transport actif des chlorures et provoquent parallèlement une natriurèse et une
perte de potassium. On range dans cette catégorie le furosémide (Lasix â).
Il convient de choisir en premier lieu un diurétique
à action distale en vue de réduire l’hyperaldostéronisme. L’Aldactone sera très
souvent efficace pour autant que la fonction rénale soit normale. La dose
initiale est de 100 mg par jour. L’action peut ne se manifester qu’après 2 ou 3
jours. Les doses ne sont donc augmentées que tous les 3 jours. Dans certains
cas, une dose de 400 mg/j peut être indispensable. Des substituts de potassium
ne sont pas indiqués au cours du traitement à l’Aldactone. En cas d’échec, il
convient d’ajouter du furosémide à doses croissantes en commençant par 40 mg
tous les deux jours. Cette dose peut être augmentée dans les cas rebelles
jusqu’à 120 mg/j. Si le traitement donne un résultat positif, des instructions
sont données au patient pour qu’il surveille son régime et son poids
régulièrement. La restriction sodée peut être moins stricte à partir du moment
où le traitement se révèle efficace. Il faut régulièrement ajuster la dose de
diurétiques.
2. Complications du traitement médical
Les effets secondaires du
traitement diurétique sont dus principalement aux désordres
hydro-électrolytiques et de l’équilibre acido-basique qu’ils peuvent engendrer.
Ils peuvent être le point de départ d’une encéphalopathie ou d’une insuffisance
rénale.
3. Ascite réfractaire
En général, la cirrhose est
sévère. Les paracentèses (vidange
d’ascite) associées à des perfusions d’albumine sont indiquées dans des
ascites importantes et réfractaires, lorsque le traitement aux diurétiques se
révèle inefficace ou contre-indiqué.
CHAPITRE IV :
LES HEPATITES VIRALES
Six virus spécifiquement responsables d’hépatites
(virus hépatotropes) sont actuellement connus (A, B, C, D, E, G).
D’autres virus du groupe de l’herpès peuvent
occasionnellement provoquer une hépatite : mononucléose infectieuse,
cytomégalovirus, herpès. La fièvre jaune est une affection virale d’origine
tropicale.
I. ANATOMOPATHOLOGIE
D’un point de vue morphologique, l’hépatite virale
est une affection nécrosante qui après une phase initiale aiguë peut guérir
avec ou sans séquelles ou évoluer sur un mode chronique.
A. HEPATITES AIGUES
Le tableau combine une atteinte diffuse des
hépatocytes et une infiltration inflammatoire à prédominance de cellules
mononucléées.
L’atteinte
hépatocytaire
peut se manifester par une ballonisation des cellules dont le contour
s’arrondit tandis que le cytoplasme et le noyau se clarifient et par une
nécrose acidophile qui conjugue un cytoplasme acidophile et un noyau pycnotique
ou karyorrectique. Certaines cellules nécrotiques se transforment en corps
acidophiles ovoïdes que l’on trouve dans les lames hépatocellulaires et dans
les sinusoïdes (corps de Councilman). La nécrose est souvent focale et
prédomine en périphérie de l’acinus. Elle peut s’étendre et devenir confluante.
Concomitamment à la nécrose, se manifestent des phénomènes de régénération sous forme de mitoses, cellules
plurinucléées et d’hépatocytes basophiles, de petite taille, qui en s’empilant
forment des lames pluristratifiées.
La réaction
inflammatoire est essentiellement mononucléée, constituée de lymphocytes et
de macrophages. Elle s’étend d’une manière diffuse dans l’acinus mais est plus
marquée aux endroits de nécrose focale. Les espaces portes sont entrepris. Ils
sont élargis, de contour irrégulier. Les cellules de Kuppfer sont
hyperplasiques et montrent des images de phagocytose.
La cholostase survient tardivement dans l’évolution.
Elle se manifeste par des thrombus biliaires.
B. HEPATITE FULMINANTE
La nécrose hépatique massive
entraîne une destruction étendue du parenchyme s’accompagnant de collapsus de
la trame de réticuline. Les lésions sont irrégulièrement dispersées dans le
foie, respectant certaines zones. Les foyers nécrotiques peuvent former des
ponts entre les structures portes et centrolobulaires voisines (bridging
necrosis) ou s’étendre à des lobules entiers voire à plusieurs lobules contigus
(nécrose multilobulaire).
C. HEPATITES CHRONIQUES
1. Hépatites chroniques en général
Environ 10% des hépatites virales B et 50 à 80%n des
hépatites virales C peuvent évoluer vers un stade chronique. Quel que soit
l’agent étiologique, l’image reste similaire et se caractérise par une atteinte diffuse d’un processus
inflammatoire nécrosant et fibrosant. Les lésions se concentrent sur les
espaces portes et la zone péri-portale. L’atteinte lobulaire est plus
inconstante.
La classification actuelle des hépatites chroniques
est basée sur :
·
Le
grande de la nécro-inflammation
(absent, modérée, sévère).
·
Le
stade de la réaction fibreuse
(absente, modérée, sévère).

Figure 12
2. Aspect particulier aux hépatites B et C
Dans l’hépatite B on peut observer des modifications d’un certain nombre d’hépatocytes dont tout ou partie du cytoplasme se clarifie en prenant un aspect granulaire (image en verre pilé). Cet aspect est lié à des modifications du réseau microtubulaire et à l’accumulation d’antigènes viraux (antigènes de surface de l’hépatite B). On peut mettre ces derniers en évidence par des réactions immuno-cytochimiques.
Les lésions de l’hépatite C se
distinguent par la fréquence de l’atteinte lobulaire, par une distribution
segmentaire et focale de l’inflammation portale avec formation d’agrégats lymphoïdes
portaux, par une stéatose modérée et par des lésions de petits canaux
biliaires.
D. CIRRHOSE POST NECROTIQUE
Certaines hépatites chroniques actives évoluent
rapidement ou insidieusement vers une cirrhose. Celle-ci se caractérise par une
fibrose mutilante.
Les nodules de régénération sont souvent plus
volumineux que dans la cirrhose alcoolique.
II. EPIDEMIOLOGIE ET EVOLUTION SEROLOGIQUE
A. VIRUS
A (Virus à ARN)
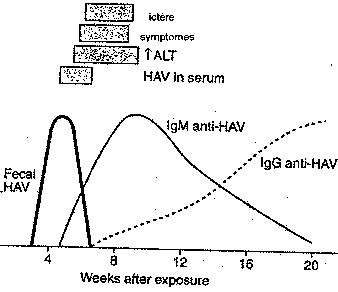 La transmission se fait par
voie féco-orale par l’eau ou les
aliments (salades, fruits, mollus-ques, huîtres) contaminés par des matières
fécales. Avec l’amélioration des conditions socio-économiques le pourcentage de
sujets adultes ayant rencontré le virus A diminue : il est actuellement
inférieur à 50%.
La transmission se fait par
voie féco-orale par l’eau ou les
aliments (salades, fruits, mollus-ques, huîtres) contaminés par des matières
fécales. Avec l’amélioration des conditions socio-économiques le pourcentage de
sujets adultes ayant rencontré le virus A diminue : il est actuellement
inférieur à 50%.
La période d’incubation est de 15 à 45 jours (moyenne 4 semaines). La virémie
est de trop courte durée et d’importance trop faible pour être détectable de
façon routinière. La présence de virus dans les selles atteint un maximum et
diminue très rapidement avant même que les symptômes cliniques ne soient
apparus.
Figure 13
La présence d’anticorps
Anti-HAV (Hépatite A Virus) dans le sérum peut être observée entre la phase
aiguë et la phase de convalescence. Alors que dans d’autres affections virales,
l’ascension du titre survient endéans deux à trois semaines, dans l’hépatite A
le laps de temps peut être plus long (4 à 6 semaines).
La détermination des deux classes d’anticorps
(anti-HAV-IgM et anti-HAV-IgG) permet d’établir avec plus de précision le stade
d’évolution de la maladie. En effet, les anticorps de la classe IgM surviennent
environ vers la 4e semaine. Dans les semaines qui suivent, ce rapport va
diminuer et après quelques semaines (8 à 12), seuls les anticorps de la classe
IgG sont prédominants, persistent ad vitam et sont associés à une immunité à
long terme.
B. VIRUS
B (Virus à ADN)
La transmission
se fait essentiellement par voie
sexuelle ou par le sang :
·
Percutanée via le sang ou les dérivés
du sang (fibrinogène, facteurs anti-hémophiliques) : transfusion, hémodialyse,
seringue des toxicomanes, contact d’une érosion cutanée avec du sang contaminé
(personnel médical et paramédical), matériel insuffisamment stérilisé
(tatouages, soins dentaires).
·
Sexuelle : homo et hétérosexuels.
·
Périnatale ou verticale : lors de l’accouchement
d’une mère infectée.
·
Familiale ou horizontale : entourage proche d’un
sujet infecté.
La période d’incubation varie de 30 à 180 jours et est, en moyenne, de 75 jours.
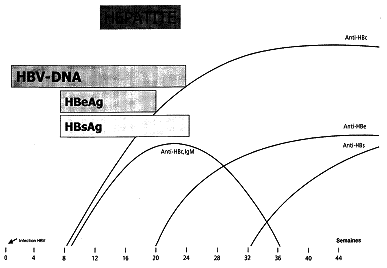 Trois antigènes du virus de
l’hépatite B ont été mis en évidence :
Trois antigènes du virus de
l’hépatite B ont été mis en évidence :
·
l’antigène
de surface : HBsAg.
·
L’antigène
nucléaire : HBcAg (« core » antigen ; antigène de nucléocapside).
·
L’antigène
e : HBeAg (sous unité de l’HBcAg).
L’HBcAg reste dans le foie, les deux autres pouvant
se trouver dans le sang. A chacun de ces trois antigènes correspond un
anticorps (anti-HBs, anti-HBc, anti-HBe). Tous trois sont utilisés en routine
clinique.
Figure 14a
|
HEPATITE |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
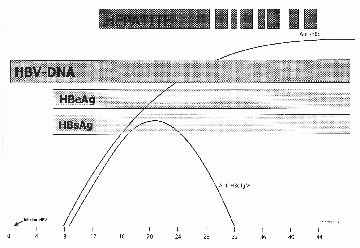
Figure 14b
L’HBsAg apparaît dans le sérum de 2 à 8 semaines avant le début
clinique et biologique de la maladie. Au cours d’une hépatite simple il
disparaît en général après quelques semaines, mais il persiste durant des mois
voire des années chez 10% des malades. Des anticorps antiHBs apparaissent dans le sang 4 à 5 mois après. l’infection.
Ils atteignent un taux maximum vers le 12ème mois
puis décroissent progressivement en plusieurs années. Leur apparition peut être
considérée comme un signe de bon
pronostic témoignant de la guérison de l’infection. L’anti-HBs peut aussi
apparaître, sans infection préalable, après immunisation active (vaccination).
L’antigène
HBe apparaît en même temps que l’HBsAg mais sa présence est plus fugace. Il
témoigne d’une phase de réplication virale et indique que le malade est
contagieux. L’anticorps anti-HBe apparaît quelques semaines après le début de la
maladie dans les cas favorables mais pas dans les infections chroniques.
L’antigène HBc n’est pas présent dans le sérum. L’anticorps anti-HBc apparaît plus
précocement que l’anti-HBe. Les IgM anti-HBc disparaissent en quelques
semaines, alors que les IgG persistent durant des années. L’anti-HBc est absent
chez les sujets vaccinés.
L’ADN viral B,
détectable dans le sang, est considéré comme un marqueur de réplication virale
et donc d’infectivité.
Le virus B mutant est caractérisé par une mutation
au niveau de la région pré-core avec incapacité de sécréter l’AgHBe mais
poursuite de la réplication virale (ADN positif).
Le tableau suivant résume la signification des
divers marqueurs de l’hépatite B.
|
INTERPRETATION DES MARQUEURS DE
L’HEPATITE B |
|||||||
|
HBsAg |
HBeAg |
HBcAc |
HBcAc IgM |
HBeAc |
HBsAc |
Interprétation |
|
|
- |
- |
- |
- |
- |
- |
Absence de contact avec le
virus |
|
|
+ |
+ |
+ |
+ |
- |
- |
Hépatite aiguë (début). |
|
|
+ |
- |
+ |
+ |
+ |
- |
Hépatite aiguë (fin). Non
contagieuse |
|
|
- |
- |
+ |
- |
+ |
- |
Hépatite aiguë en voie de
guérison |
|
|
- |
- |
+ |
- |
+ |
+ |
Antécédents d’hépatite B |
|
|
- |
- |
- |
- |
- |
+ |
Etat post vaccination |
|
|
+ |
+ |
+ |
+ ou - |
- |
- |
Hépatite chronique (début) |
|
C. VIRUS
d (virus à ARN)
C’est un virus défectif, dépendant du virus B pour
sa réplication.
La transmission peut se faire de personne à personne
ou par voie parentérale. Cette transmission se fait soit à l’occasion d’une
infection simultanée avec le virus B (co-infection) soit lors d’une
surinfection chez un malade porteur du virus B.
Au cours de l’infection aiguë l’antigèned est détecté transitoirement, suivi de
l’anticorps d. Au cours de l’infection
chronique, les meilleurs marqueurs sont l’anti d sérique et l’antigène d dans le foie.
D. VIRUS
C (virus à ARN)
La transmission se fait par le sang ou par du
matériel souillé par du sang contaminé.
·
Percutanée par le sang ou les dérivés
du sang, les aiguilles contaminées (toxicomanes) ou par des piqûres
accidentelles.
·
Sexuelle. Le risque est moins grand
que pour le virus B (de l’ordre de 5%).
·
Interfamiliale horizontale via du matériel contaminé
par du sang infecté.
·
Verticale de la mère à l’enfant (de
l’ordre de 5%).
·
Inconnue (40%).
La durée de la période
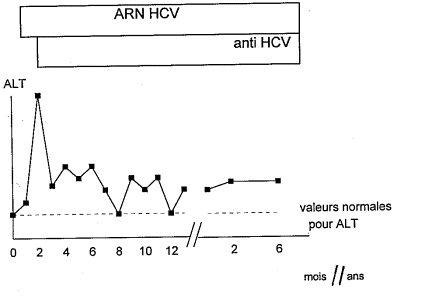
Figure 15
Le diagnostic du virus C se fait par la recherche de
l’anticorps anti-VHC. Ce test ne se
positive qu’après 2 mois et est donc rarement présent au stade aigu.
L’apparition d’anticorps anti VHC ne témoigne pas de la guérison de
l’infection.
L’ARN du virus C peut être détecté par PCR dans le
sang et signe une réplication virale. Cette mesure est utile pour le diagnostic
d’une hépatite aiguë C (avant la positivation des anti VHC) ou pour le suivi de
l’efficacité du traitement anti-viral.
E. VIRUS E
Le virus de l’hépatite E est
transmis par voie féco-orale comme
le virus de l’hépatite A. Il est responsable d’épidémies principalement en Asie et au Moyen Orient.
F. VIRUS G
Le virus de l’hépatite G est
transmis par voie sanguine et apparaît peu pathogène.
G. AUTRES VIRUS
1. Mononucléose infectieuse (EBV)
Cette
maladie est provoquée par le virus herpétique d’Epstein-Barr (EBV). Elle se
présente surtout chez les adolescents et les jeunes adultes. Elle est associée
à une augmentation des anticorps IgM contre les antigènes viraux
d’Epstein-Barr. La réaction de Paul et Bunnel garde une grande valeur
diagnostique. L’atteinte hépatique de la mononucléose infectieuse est
généralement modérée et ressemble à celle des autres hépatites virales. Un
ictère n’est présent que dans 10% des cas, une splénomégalie dans 50% des cas.
2. Cytomégalovirus (CMV)
Ce virus peut être isolé de
l’urine et de la salive et peut être transmis par transfusion sanguine.
L’atteinte hépatique peut n’être qu’un élément d’une maladie diffuse chez les
patients immuno-déprimés. On observe une augmentation du taux d’anticorps
fixant le complément ainsi que des anticorps anti-IgM.
3. Fièvre jaune
Cette infection endémique en
Amérique du Sud et en Afrique Centrale est due à un arbovirus du groupe B et
est transmise par des moustiques infectés. D’autres hépatites virales de ce
type existent (fièvre de Lhassa, fièvre d’Ebola, etc...).
|
DIAGNOSTIC ETIOLOGIQUE DE L’HEPATITE VIRALE AIGUE |
|
|
VIRUS |
MARQUEURS VIRAUX |
|
Hépatite A Hépatite B Hépatite B delta (d) Hépatite C Hépatite EBV Hépatite CMV |
anti HA IgM HBsAg,
Anti-HBc IgM Ag d, anti d Anti VHC (après 2 mois) Paul et Bunnel, anti-EBV IgM Lymphocytose, lymphocytes atypiques Anti-CMV IgM Lymphocytes atypiques |
III. MANIFESTATIONS CLINIQUES
-
Dans
80% des cas les hépatites virales aiguës sont asymptomatiques.
-
Lorsqu’elles
sont symptomatiques, le tableau est assez stéréotypé et identique pour tous les
virus responsables.
Le tableau comprend alors trois phases.
1. Phase pré-ictèrique
Durant quelques jours (2 à
15 jours) le patient se plaint de troubles peu spécifiques : asthénie, anorexie, dégoût des graisses.
Une douleur épigastrique est
fréquemment notée, parfois assez intense au point de prêter à confusion avec
une douleur d’origine vésiculaire. La fièvre est, en général, modérée mais une
fièvre augmentant brutalement en intensité (39,5°) peut se présenter durant 1 à
4 jours. Des arthralgies sont
souvent présentes ; un rash de type
urticarien est plus rare.
2. Phase ictérique
La durée
de cette phase est très variable. Si, en général, elle est de 2 à 6 semaines, elle peut persister
dans de rares cas pendant plus de 3 mois. Les symptômes de la phase
préictérique ont tendance à s’amender sauf de l’asthénie. L’ictère atteint son maximum en quelques jours au-delà desquels
l’appétit redevient normal. Une hépatosplénomégalie peut s’observer. Un
amaigrissement se manifeste durant les premières semaines. Les selles sont plus ou moins décolorées
avec éventuellement stéatorrhée.
3. Phase post-ictérique
Parallèlement au retour de la bilirubinémie à un
taux normal, l’ensemble de la symptomatologie disparaît progressivement. Le
symptôme le plus rebelle est l’asthénie qui peut persister longtemps.
D’autres formes cliniques peuvent exister durant la
phase aigüe avec une évolution soit favorable - formes anictériques, cholostatiques ou
récidivantes (surtout pour l’hépatite A), formes avec manifestations extra-hépatiques (atteintes
neuro-musculaire, hématologique, rénales, rhumatologiques, dermatologiques)- soit
défavorable : hépatite aiguë
sévère (avec chute du PTT inférieure à 50%) pouvant déboucher sur une
hépatite fulminante ou sub-fulminante dans 1% des cas (voir chapire
insuffisance hépatique aiguë).
IV. DIAGNOSTIC BIOLOGIQUE
La bilirubine
est élevée dans les formes ictériques.
L’anomalie la plus importante est l’augmentation du
taux des transaminases qui est
précoce et précède en générale l’ictère. Au cours de l’évolution, elle dépasse souvent de 10 fois la limite
supérieure de la normale. Sa régression est variable : des taux légèrement
anormaux peuvent persister durant quelques mois après disparition de l’ictère.
Le taux de la phosphatase
alcaline est normal ou légèrement élevé (2x la normale) sauf dans la forme
cholostatique où elle est beaucoup plus élevée.
La ¡ GT est élevée (en général 5 à
10 fois la normale).
La présence des marqueurs
viraux (voir tableau) précédemment décrits est un élément important du
diagnostic des hépatites virales et aide grandement au diagnostic différentiel
des cytolyses aiguës (cf. tableau).
|
CAUSES DE CYTOLYSE AIGUE |
|||
|
ALT > 10 fois la normale |
ALT < 10 fois la normale |
||
|
Causes |
Moyens diagnostiques |
Causes |
Moyens diagnostiques |
|
Hépatite virale (A, B, C, D) Hépatite médicamenteuse Hépatite autoimmune Foie hypoxique Migration lithiasique biliaire Budd-Chiari Maladie de Wilson |
Sérologie Contexte clinique – biopsie Anticorps anti-tissus Echographie Doppler – biopsie Echographie – cholangiographie (RMN, rétrograde Echographie Doppler Cu urinaire, céruloplasmine |
Hépatite virale (C, CMV, EBV) Poussée aiguë d’une hépatite chronique B Hépatite médicamenteuse Hépatite alcoolique |
Sérologie Sérologie Contexte clinique, biopsie Biopsie – biologie |
V. EVOLUTION ET PRONOSTIC
L’évolution à long terme est toujours favorable pour
l’hépatite A, l’hépatite E, la mononucléose et l’hépatite à cytomégalovirus.
Quatre types d’évolution au long courts ont été
décrits pour les hépatites B et C : les porteurs asymptomatiques du virus,
l’hépatite chronique, la cirrhose, le carcinome hépatocellulaire.
A. PORTEURS SAINS
Il s’agit de sujets porteurs du virus B ou C dans le foie, sans lésions hépatiques
associées. Les transaminases sont normales. Au niveau de la sérologie, on
observe un HBsAg positif, le plus souvent en l’absence de multiplication virale
(HBeAg et ADN VHB négatifs) ou un anti-VHC positif associé à de l’ARN viral
positif par PCR.
B. HEPATITE CHRONIQUE
Elle se définit par la
persistance d’une élévation des transaminases plus de six mois après l’épisode aigu. L’évolution vers la
chronicité s’observe dans environ 10%
des cas d’hépatite B ainsi que dans environ 50 à 80% des hépatites C. La persistance d’une inflammation
hépatique après la phase aiguë est liée soit à une réponse immunitaire
inadéquate avec impossibilité d’éliminer les antigènes viraux, soit à un
processus auto-immune cytotoxique.
La symptomatologie consiste en une lassitude persistante, une gêne de l’hypocondre droit ou du
prurit. Des arthralgies peuvent être
le symptôme prédominant. Une hépatomégalie
peut être présente.
L’anomalie
biologique
constamment retrouvée est l’élévation du taux de transaminases qui est
habituellement modérée (entre 1,5 et 5 x la normale). Les transaminases ALT sont supérieures aux AST. La bilirubine n’est
élevée et le PTT abaissé qu’en cas d’insuffisance hépatique chronique (associée
à une hépatite chronique très active ou avec une cirrhose). Au niveau sérologique, l’hépatite chronique
B et l’hépatite chronique C se caractérisent par la persistance pendant plus de
6 mois, respectivement de l’AgHBs (pour la B) et de l’anticorps anti VHC et de
la virémie C par PCR (pour la C).
L’hépatite
chronique B
évolue schématiquement en trois phases, parfois suivie d’une réactivation :
·
Une
phase de réplication virale (AgHBe +, DNA sérique +) avec une faible activité
biologique (transaminases peu élevées) et histologique pouvant durer de 5 à 10
ans (tolérance immune).
·
Une
diminution de la réplication virale avec forte activité biologique et
histologique. Au moment où intervient la séro-conversion (AgHBe ® anti HBe), des lésions irréversibles de
cirrhose peuvent être constituées chez certains patients (clairance immune).
·
Une
disparition de la réplication virale (HBeAg -, antiHBe +) ainsi que de
l’activité biologique et histologique. (Portage sans réplication virale : le
virus B est intégré).
·
Parfois,
spontanément ou à l’occasion d’un état d’immunodépression, il y a une
réactivation.
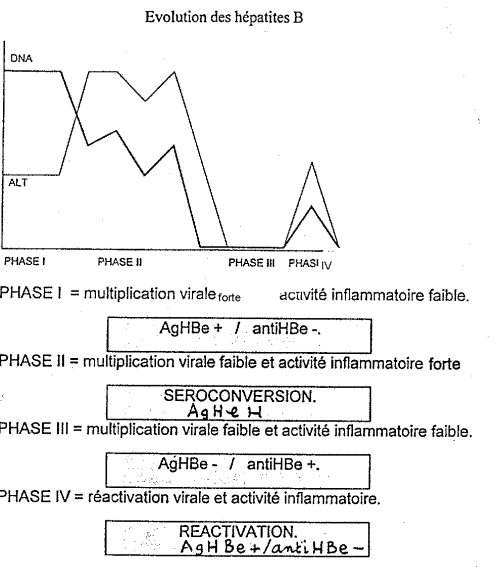
Figure 16
Le taux de séroconversion AgHBe ® anti HBe est de l’ordre de 5 à 10% par an.
L’hépatite
chronique C
est souvent asymptomatique et ne se manifeste que par une élévation modérée et
fluctuante des transaminases.
Les hépatites chroniques virales doivent être
différenciées des autres causes de cytolyse.
|
CAUSES DE CYTOLYSE CHRONIQUE |
|
|
|
Exemples |
|
Hépatopathie virale
chronique Hépatopathie auto-immune Hépatopathie
médicamenteuse Stéato-hépatite alcoolique
ou non alcoolique Hépatopathies métaboliques
et génétiques Hépatopathies vasculaires Hépatopathies
cryptogéniques |
C, B, Bd INH, Dantrolène, améthyldopa Obésité, diabète,
médicaments Hémochromatose Maladie de Wilson Déficit en a1 antirypsine Foie cardiaque Syndrome de Budd-Chiari Maladie veino-occlusive |
C. CIRRHOSE HEPATIQUE
L’évolution d’une hépatite
chronique vers une cirrhose est observée essentiellement dans deux
circonstances : les cas d’hépatites
fulminantes subaiguës au cours desquelles une cirrhose peut se développer
en quelques mois, et dans les hépatites
chroniques avec activité et fibrose importantes au cous desquelles une
cirrhose peut se développer en 10 à 20 ans.
D. HEPATOCARCINOME
Dans la grande majorité des cas, la cirrhose est une
étape préalable au développement du cancer du foie.
VI. TRAITEMENT
A. MESURES GENERALES
Le repos au lit n’est pas conseillé et il faut
éviter toute médication car leur métabolisme hépatique est diminué.
Le régime doit être équilibré.
B. MEDICAMENTS
1. Hépatite aiguë
Aucun n’est efficace en dehors de la cortisone pour
l’hépatite aiguë A cholostatique et de l’interféron a pour l’hépatite aiguë C.
2. Hépatite chronique
Le traitement a pour objectif d’arrêter la multiplication virale (interferon
alpha ou analogues nucléosidiques) afin d’éviter l’évolution vers la cirrhose
et de stimuler l’immunité cellulaire
(interferon alpha).
Il est donc logique de traiter l’hépatite virale
chronique à un stade précoce, avant l’installation d’une cirrhose.
1) Hépatite chronique B
L’interféron
alpha (10 millions d’unités, 3 fois par
semaines, durant 4 à 6 mois par voie sous-cutanée) induit une séroconversion HBe chez 40 à 50% des
patients porteurs d’une hépatite chronique active B avec multiplication virale
(Ag HBe + DNA+).
La disparition de l’antigène HBs est plus rare et plus
tardive. Les facteurs permettant d’augurer une réponse favorable au traitement
sont : un taux élevé de transaminases, un taux bas d’ADN viral,
l’hétérosexualité, le sexe féminin et l’absence d’infection par les virus HIV,
delta et B mutant.
L’interféron reproduit avec un pourcentage plus
élevé les phases 2 et 3 précédemment décrites pour l’évolution de l’hépatite B
chronique. Il est contre-indiqué dans
la cirrhose décompensée.
La lamivudine (analogue nucléosidique) prise
par voie orale est indiquée dans la cirrhose décompensée (avant
transplantation), dans l’hépatite chronique évolutive B delta et liée au virus
mutant precore.
2) Hépatite
chronique C
L’interféron a sera proposé (3 millions
d’unités 3 x par semaine) associé à la ribavirine (analogue nucléosidique) chez
les patients porteurs d’une hépatite chronique C avec activité et fibrose
significative à l’histologie et pour autant qu’il n’y ait pas de
contre-indication à ces médicaments, pendant 6 à 12 mois ce qui permet
d’obtenir une réponse soutenue (c’est-à-dire des ALT normales et une virémie
négative 6 mois après l’arrêt du traitement) dans 40% des cas (cf. figure 17).
Les facteurs augurant une réponse favorable au
traitement sont le jeune âge, l’absence de consommation excessive d’alcool, la
contamination non transfusionnelle, l’absence de cirrhose, une virémie basse et
un génotype favorable.
Hépatite
chronique C
traitée par Interferon et
Ribavirine
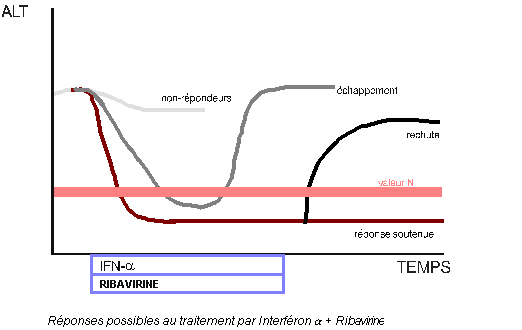
Figure 17
VII. PREVENTION
A. MESURES D’HYGIENE
Pour éviter la transmission
de l’hépatite A, les mesures habituelles sont en général inadéquates parce
qu’elles sont d’application difficile et que le virus a pu être disséminé avant
que le diagnostic soit posé (2 semaines avant l’apparition de l’ictère).
En ce qui concerne l’hépatite B et l’hépatite C, des procédures de désinfection adéquates doivent être appliquées à tout le matériel endoscopique ou potentiellement contaminant.
Il faut aussi préconiser l’usage personnel exclusif des objets contondants (rasoir, coupe-ongles, brosse à dents, ...) et discuter de l’usage du préservatif chez l’homme.
B. IMMUNOGLOBULINES ET VACCINATION
1. Hépatite A
L’administration d’immunoglobulines spécifiques associées
au vaccin (HAVRIX â) est recommandée chez les
personnes ayant été au contact d’une hépatite A pour autant qu’elle soit
réalisée le plus rapidement possible (endéans les 15 jours de l’exposition au
virus).
La vaccination
est indiquée chez les voyageurs dans des pays à haut risque (hygiène et
conditions sanitaires insuffisantes), chez les travailleurs manipulant des
denrées alimentaires, chez le personnel des crèches, garderies et services de
pédiatrie, chez les toxicomanes et les patients porteurs d’hépatopathie chronique.
Le vaccin est une souche inactivée. Deux doses sont
administrées chez l’adulte aux temps 0 et 6 à 12 mois plus tard. Un rappel doit
être fait 20 ans plus tard.
2. Hépatite B
Une prévention par l’association d’immunoglobulines spécifiques anti-HBV et d’un programme
de vaccination se justifie pour la prévention d’une transmission
périnatale, après piqûre accidentelle et après un contact sexuel avec une
personne infectée.
Une vaccination
universelle est actuellement recommandée, dans le but d’éradiquer
l’hépatite virale B dans les 15 ans à venir. Le vaccin actuellement disponible
(ENGERIX â) est très efficace. Le
schéma classique de la vaccination comporte une dose administrée en
intramusculaire dans le deltoïde aux temps 0, 1 et 6 mois. Une injection supplémentaire
sera faite à 12 mois chez les sujets à risque. Il est conseillé de suivre la
réponse des anti HBs qui doivent être supérieures à 10 mU. Un rappel est
conseillé après 5 à 10 ans. En cas de non-réponse au vaccin (obésité, patient
âgé ou immunodéprimé, facteurs génétiques), il faut répéter les doses.
CHAPITRE V :
LES HEPATITES TOXIQUES ET MEDICAMENTEUSES
I. LES HEPATITES TOXIQUES
Les lésions hépatiques produites par des substances
toxiques surviennent en général, rapidement (1 à 2 jours) après leur
administration. Elles sont reproductibles. D’autres organes que le foie peuvent
être atteints.
Les lésions observées sont très variables, distribuées de façon uniforme dans
tout le lobule allant du simple oedème cellulaire jusqu’à la nécrose cellulaire
avec ou sans réaction inflammatoire. De l’infiltration graisseuse est
fréquemment observée. Dans certains, lorsque la nécrose n’est pas fatale, on
aboutit à un état de cirrhose post-nécrotique ; pour d’autres par contre, ce
n’est que la répétition des accidents toxiques qui aboutit à cette situation.
A. L’ALCOOL
L’intoxication éthylique aiguë, en dehors d’un
éthylisme chronique, ne retentit presque jamais sur le foie. Par contre,
l’éthylisme chronique conduit à des lésions hépatiques allant de la stéatose à
la cirrhose (voir stéastoses et cirrhoses).
B. L’AMANITE PHALLOIDE
Le toxique responsable des lésions et particulièrement de l’hépato-toxicité est la phalloïdine qui a pu être isolée par chromatographie.
Dans les 12 heures qui suivent l’ingestion, on assiste à l’apparition de nausées, de crampes abdominales, de diarrhée parfois sanglante et de déshydratation. Des lésions rénales, hépatiques et éventuellement du système nerveux central apparaissent dans les jours suivants, parfois après une amélioration.
Les lésions hépatiques comportent une destruction cellulaire massive pouvant entraîner le décès en l’absence de transplantation hépatique.
Le tableau clinique des
manifestations cliniques est celui d’une cytolyse avec insuffisance hépatique
modérée à très sévère.
C. LES SOLVANTS INDUSTRIELS
Les hydrocarbures halogénés
inhalés ou ingérés accidentellement ou « sniffés » peuvent déterminer
une hépatite cytolytique.
II. LES HEPATITES MEDICAMENTEUSES
Les 3 étapes du métabolisme des médicaments sont :
(1) La captation par les hépatocytes.
Certains médicaments sont avidement extraits du sang (à plus de 70%) par le foie au premier passage.
Leur
clairance est donc essentiellement fonction du flux sanguin hépatique. C’est le
cas de la morphine, du propranolol et du Vérapamil dont la dose par prise doit
être réduite en cas de cirrhose ou de shunt portosystémique spontané ou
chirurgical. Certaines de ces substances à haute extraction sont utilisées pour
l’évaluation du flux sanguin hépatique (dextropropoxyphène, lidocaïne, vert
d’indocyanine).
D’autres médicaments ont une extraction faible (< 30%). Leur clairance est
indépendante du flux sanguin hépatique mais dépend de la fonction métabolique
de l’hépatocyte. C’est le principe de l’évaluation de la fonction hépatique par
le test respiratoire à l’aminopyrine C14.
(2) La biotransformation dans l’hépatocyte.
Deux mécanismes sont possibles :
· une oxydation au niveau du cytochrome P450 (= microsome) avec formation d’un métabolite réactif. L’activité microsomiale peut être augmentée par d’autres médicaments ou toxiques (alcool, barbituriques, rifampicine, diphénylhydantoïne) ou diminuée (mutation génétique, kétoconazole, cimétidine).
· une conjugaison de la substance mère ou du métabolite réactif à une molécule endogène (acide glucuronique, glutathion, acide aminé).
(3) L’élimination du médicament hors de l’hépatocyte vers le sang ou la bile.
L’excrétion
biliaire est la plus fréquente.
Deux types d’hépatotoxicité médicamenteuse sont
possibles lorsque les mécanismes de conjugaison sont dépassés ou lorsque les
mécanismes d’induction microsomiale sont exacerbés. L’une correspond à une toxicité prévisible (liée à la dose)
par fixation du métabolite réactif sur les protéines cellulaires. L’autre
correspond à une toxicité imprévisible
(non liée à la dose) par fixation du métabolite réactif sur les protéines
cellulaires ou membranaires, formation d’un néoantigène ou haptène et réaction
immuno-allergique avec parfois apparition de symptômes d’hypersensibilité
(fébrilité, rash, éosinophilie, adénopathies).
Selon le type d’action et l’endroit où elle
s’exerce, les lésions hépatiques provoquées seront différentes. Elles
recouvrent un large spectre résumé dans le tableau suivant.
|
LESIONS HEPATIQUES INDUITES PAR LES MEDICAMENTS |
|
|
|
Exemples |
|
ATTEINTE AIGUE Hépatite aiguë Cytolitique Cholostatique Granulomateuse Surcharges Stéatose macrovacuolaire Stéatose microvacuolaire Lésions de type alcoolique ATTEINTE CHRONIQUE Hépatite chronique / cirrhose Cholostase / cirrhose biliaire Cholangite sclérosante LESIONS VASCULAIRES Sinusoïdales, centro-lobulaires Veines sus hépatiques Péliose hépatique LESIONS TUMORALES Adénomes, hépatome Angiosarcome |
INH, kétoconazole, paracétamol Erythromycine, contraceptifs oraux, ac.
Clavulanique Allopurinol, sulfaméthoxazole Corticoïdes, methotrexate Acide valproïque Amiodarone Méthyldopa, dantrolène Chlorpromazine, carbamazépine FUDR, formol Vitamine A, méthotrexate, azatioprine, arsenic Contraceptifs oraux Stéroïdes anabolisants et androgéniques Contraceptifs oraux, stéroïdes anabolisants et
androgéniques, chlorure de vinyle. |
CHAPITRE VI :
LES STEATOSES ET LES STEATOHEPATITES
I. ALCOOLIQUES
A. HISTOIRE NATURELLE DU FOIE ALCOOLIQUE
L’intoxication chronique aboutit au développement de
lésions hépatiques dont le terme ultime est la cirrhose. Le risque de
développement de lésions hépatiques est proportionnel à la quantité totale
d’alcool consommée durant la vie. La
dose seuil est de l’ordre de 210 g d’alcool par semaine chez l’homme et de 140
g par semaine chez la femme (10 g d’alcool = 1 verre de vin, 1 verre de
bière, 1 verre de whisky). Le risque d’évolution vers la cirrhose est lié à la
quantité d’alcool quotidiennement (60 g pour l’homme, 40 g pour la femme) et à
la durée de l’intoxication (10 à 20 ans).
On peut très schématiquement séparer trois entités
anatomo-cliniques :
·
La
stéatose hépatique, qui se caractérise par une surcharge lipidique des
hépatocytes. Elle peut apparaître rapidement et est réversible au sevrage.
Après un certain temps d’évolution, une fibrose apparaît (stéato-fibrose).
·
L’hépatite
alcoolique est caractérisée par des lésions hépatocytaires.
·
La
cirrhose alcoolique caractérisée par un bouleversement de l’architecture
hépatique. La stéatose ne joue aucun rôle dans le développement des cirrhoses.
C’est l’hépatite qui constitue l’étape indispensable.
Ces 3 types de lésions sont le plus souvent
associées à des degrés divers chez le même individu.
B. ANATOMOPATHOLOGIE
1. La stéatose et la stéato-fibrose
L’alcool éthylique est susceptible d’entraîner une
surcharge lipidique du cytoplasme hépatocellulaire (stéatose). Les graisses
accumulées correspondent en majorité à des triglycérides.
La stéatose prédomine dans la zone périphérique (zone 3) de l’acinus. Les
lipides s’accumulent en une large vacuole, unique, refoulant le noyau (stéatose macrovacuolaire). La
coalescence de macrovacuoles par rupture des parois cellulaires amincies peut
provoquer la formation de kystes graisseux.
2. L’hépatite alcoolique
L’hépatite alcoolique peut survenir à n’importe quel
stade de l’alcoolisme chronique. Elle se caractérise par une combinaison
d’atteinte hépatocellulaire, d’inflammation et de fibrose.
L’atteinte
hépatocytaire
se marque par une ballonisation cellulaire pouvant s’accompagner de nécrose
acidophile. Certaines cellules lésées renferment dans leur cytoplasme des
formations acidophiles, hyalines, de contours irréguliers, géographiques,
parfois annulaires. En microscopie électronique, ces corps de Mallory sont
constitués d’un agrégat de microfilaments et de substance granulaire ou
amorphe. On les observe dans 2/3 des cas d’hépatite alcoolique. Leur
spécificité est loin d’être parfaite puisqu’on peut les rencontrer dans
d’autres conditions pathologiques (cholostase de longue durée).
La réaction
inflammatoire est essentiellement constituée par des polynucléaires qui
s’accumulent autour des cellules lésées (satellitose)
et dans les sinusoïdes adjacents. Les cellules mononucléées sont relativement
rares et se rencontrent plutôt dans les cas légers ou lors de la résolution de
l’hépatite. L’inflammation portale est, en général, peu marquée.
La fibrose
constitue un paramètre constant. Elle prédomine dans la zone périphérique de
l’acinus. Elle se présente sous deux aspects histologiques imbriqués. La
fibrose péri-cellulaire se développe par dépôt de collagène dans l’espace de
Disse suite à une transformation fibroblastique des cellules de Ito. Elle
entraîne une capillarisation des sinusoïdes qui entrave les échanges
métaboliques. La fibrose périveinulaire se développe dans la paroi et autour
des veines centrolobulaires.
Pour de nombreux auteurs, la fibrose est toujours la
conséquence de phénomènes de nécrose inflammatoire (hépatite). Pour d’autres,
elle peut s’observer sur un foie stéatosique, indépendamment de toute nécrose
(stéato-fibrose).
Dans les stades avancés de l’hépatite alcoolique, la
fibrose prédomine. Elle réalise des ponts et septa disséquant le parenchyme.
Les efforts de régénération cellulaire aboutissent à la formation de nodules
ayant perdu l’architecture lobulaire. A ce stade, la distinction histologique
avec une cirrhose est ténue. On utilise parfois le terme d’hépatite en
transformation cirrhotique.
3. La cirrhose alcoolique
Cf. le chapitre « cirrhose ».
C. MANIFESTATIONS CLINIQUES, DIAGNOSTIC, TRAITEMENT
1. La stéatose
Le seul symptôme est la présence d’un gros foie mou, lisse, parfois douloureux.
Il existe des signes biologique de cytolyse modérée et/ou de cholostase. Une hyperlipémie avec forte
élévation des triglycérides s’accompagnant parfois d’une anémie hémolytique
peut être observée (syndrome de Zieve).
En laparoscopie le foie est clair, jaune chamois
lisse. Une biopsie confirme le diagnostic.
2. L’hépatite alcoolique
Les signes cliniques habituels sont : des douleurs de l’hypochondre droit, des nausées et des vomissements, un ictère et un syndrome fébrile. Des signes d’aggravation d’une hépatopathie
préexistante peuvent apparaître (ascite, encéphalopathie, hémorragies
digestives).
Les examens
biologiques montrent : une hyperleucocytose
à polynucléaires neutrophiles, une anémie
macrocytaire modérée, une augmentation
modérée des transaminases avec un rapport AST/ALT supérieure à 1, une hyperbilirubinémie conjuguée, une élévation des immunoglobulines (en
particulier les IGA). Une élévation des phosphatases alcalines et des gammas GT
est plus rare.
L’échographie ne montre pas d’anomalies
caractéristiques, mais peut révéler la présence d’une stéatose d’une cirrhose
ou d’une hypertension portale.
La biopsie
hépatique apporte un diagnostic de certitude.
Le diagnostic différentiel doit être fait entre une
hépatite alcoolique, une hépatite virale, une angiocholite ou une hépatite
médicamenteuse.
En dehors de la suppression totale des boissons
alcooliques et du traitement de l’ascite et des troubles de l’hémostase, peu de
thérapeutiques sont efficaces. Les corticostéroïdes réduisent la mortalité des
hépatites alcooliques graves et sont préconisés pour autant qu’il n’y ait pas
d’infection.
II. NON ALCOOLIQUES
La stéatose non alcoolique peut se présenter sous
deux formes : macrovacuolaire et microvacuolaire.
1. Forme macrovacuolaire
Les lésions hépatiques sont semblables à celles
observées dans la stéatose alcoolique.
En dehors de l’alcoolisme, la cause la plus fréquente
de stéatose macrovacuolaire, dans les pays industrialisés, est l’excès pondéral. L’intensité de la
stéatose n’est pas proportionnelle à l’obésité : on observe des stéatoses
importantes chez des malades dont l’excès pondéral est modeste.
Les causes plus rares sont le diabète, la carence
protidique et la malnutrition, l’hyper-corticisme, l’administration de
corticoïdes et la nutrition parentérale.
La stéatose
macrovacuolaire ne provoque en général aucun symptôme.
Les tests hépatiques sont souvent normaux, mais on
peut observer une augmentation modérée des transaminases, des phosphatases
alcalines et des ¡ GT. La stéatose par excès
pondéral est responsable de la moitié des cas d’augmentation asymptomatique des
transaminases dans les pays industrialisés.
L’échographie montre un foie hyperéchogène de
manière homogène ou hétérogène. A la tomodensitométrie le foie est hypodense.
Ces modifications ne sont pas constantes et dans certains cas la biopsie est
nécessaire pour confirmer le diagnostic.
2. Forme microvacuolaire
La cellule prend un aspect spumeux à cause de la
présence de vésicules optiquement vides dans les préparations classiques. Le
noyau reste central.
Parmi les causes de stéatose microvacuolaire on peut
citer l’administration de médicaments et la stéatose gravidique.
Ce type de stéatose entraîne un dysfonctionnement de
l’hépatocyte modéré ou sévère, éventuellement mortel.
Le tableau clinique est en général dominé par
l’ictère. Il existe une augmentation des transaminases, des phosphatases alcalines
et des ¡ GT.
|
LES STEATOSES HEPATITES NON ALCOOLIQUES |
|
|
Stéatose macro-vacuolaire Stéatose micro-vacuolaire |
Obésité, boulimie Alcool Diabète gras Médicaments : corticoïdes,
méthotrexate, amiodarone Hépatite C Carences protidiques Nutrition parentérale Hypertriglycéridémie Médicaments : Acide valproïque, AINS, Antiviraux (DDI, AZT) Alcool Stéatose gravidique Syndrome de Reye |
CHAPITRE VII :
PATHOLOGIE HEPATIQUE AUTO-IMMUNE
I. LES HEPATITES AUTO-IMMUNES
Les hépatites auto-immunes sont la conséquence d’une
réaction auto-immune contre des constituants de la membrane des hépatocytes.
Les hépatites auto-immunes de type I sont caractérisées par des anticorps antinucléaires et/ou des anticorps anti-muscles lisses (de type actine) à un titre élevé.
Les hépatites auto-immunes de
type II sont caractérisées par des anticorps anti-LKM à un titre élevé. Les
antigènes correspondants sont des enzymes du cytochrome P450 du réticulum
endoplasmique des hépatocytes et de l’épithélium des tubes contournés du rein.
La maladie affecte principalement les femmes soit entre 20 et 30 ans soit à la
ménopause.
Les manifestations cliniques sont généralement superposables à celles des hépatites
virales aiguës ou chroniques. Ce type d’hépatite est néanmoins plus
fréquemment associé à d’autres maladies
auto-immunes (thyroïdite d’Hashimoto, arthrite rhumatoïde, Maladie de
Sjörgen). On met fréquemment en évidence le
facteur antinucléaire, des anticorps anti-muscle lisse, ou des anticorps anti
LKM (liver kidney microsomes) ainsi qu’une hypergamma-globulinémie.
La biopsie hépatique montre une infiltration
inflammatoire lymphoplasmatocytaire des espaces portes avec « peace
meal necrosis » et une fibrose plus ou moins marquée.
L’évolution de ce type d’hépatite chronique est fluctuante et est marquée par des
épisodes répétés de détérioration aboutissant le plus souvent à une cirrhose.
Si le diagnostic différentiel avec l’hépatite chronique B est aisé par
l’évaluation des marqueurs sérologiques, la confusion est possible avec une hépatite
chronique C dont on connaît également le caractère fluctuant. L’utilisation du
marqueur spécifique de l’hépatite C (anti HCV) est un apport important au
diagnostic différentiel.
Le principe du traitement consiste à administrer
durant une période prolongée une association de prednisolone et d’azathioprine
après biopsie hépatique démontrant le caractère actif de la maladie. Les
indications de ces traitements au long cours, dont on connaît les effets
secondaires non négligeables, doivent être pesées en fonction du contexte
clinique et de la gravité des symptômes.
II. LA CIRRHOSE BILIAIRE PRIMITIVE
La cirrhose
biliaire primitive est une inflammation chronique des petites voies
biliaires intra-hépatiques responsables d’une cholostase chronique. Une cirrhose
caractérisée ne se développe qu’après de nombreuses années d’évolution et le
terme de cirrhose est donc mal approprié.
La pathogénie de cette affection reste imprécise ;
elle est vraisemblablement liée à des troubles
immunitaires sous la dépendance de facteurs génétiques et infectieux.
Dans la grande majorité des cas (90%), la maladie
survient chez la femmes entre 35 et 55
ans. Après une phase asymptomatique de durée variable (parfois plusieurs
années), un prurit et un subictère
se manifestent. Le prurit peut précéder l’ictère de plusieurs mois. Ce n’est
qu’après plusieurs années que l’ictère s’intensifie et que les conséquences
classiques de la cholostase sont
observées : malabsorption des graisses, ostéoporose et ostéomalacie, xanthomes
et xanthélasma. Une association avec d’autres
affections auto-immunitaires (maladie de Sjörgen, syndrome de Raynaud,
thyroïdite) n’est pas exceptionnelle.
L’examen physique est généralement banal au début de
l’évolution. Une hépatosplénomégalie
peut se développer ultérieurement de même que des signes d’hypertension portale et d’insuffisance
hépato-cellulaire (ascite, oedèmes des membres inférieurs, hémorragies
cutanées ou digestives, ictère).
Le diagnostic précoce de la maladie à la phase
asymptomatique repose sur une augmentation du taux de la phosphatase alcaline, de la ¡GT et du taux
des IgM.
Des anticorps
antimitochondriaux (anti M2) sont présents chez plus de 50% des malades avant
l’apparition des signes cliniques. Leur fréquence est de plus de 90% à des
stades plus évolués de la maladie.
La biopsie hépatique montre une cholangite
destructive avec infiltration à mononucléaires au niveau des espaces portes et
un degré variable de fibrose portale allant jusqu’à la cirrhose.
Au cours de l’évolution, le taux de bilirubinémie, resté stable durant
plusieurs années, augmente progressivement et est annonciateur de la survenue
de la dernière phase de la maladie. Parallèlement, le taux de cholestérol
s’élève progressivement et ne diminue qu’en fin d’évolution, en présence d’une
insuffisance hépato-cellulaire.
La cholostase étant démontrée, il faut différencier
celle-ci des autres causes de cholostase. Une cholangiographie rétrograde
endoscopique est souvent indiquée. Une biopsie
hépatique est indispensable pour confirmer le diagnostic et pour évaluer la
sévérité de la maladie.
L’évolution fatale est la règle après 10 à 15 ans
d’évolution.
Actuellement, il n’y a pas de traitement susceptible
de modifier le cours évolutif de la maladie vers la cirrhose. Le traitement est
symptomatique. On recommande
l’administration d’acide ursodésoxycholique
en se basant sur la notion de cytotoxicité biliaire des acides biliaires par
effet détergent. Celle-ci dépend de la proportion hydrophobique/hydrophilique
de chaque sel biliaire. Elle est maximale pour le lithocholate et minimale pour
l’acide ursodésoxycholique. L’acide ursodésoxycholique diminue le taux des
acides biliaires cytotoxiques. Son administration à la dose de 10 à 20 mg/kg
entraîne rapidement une diminution du taux des enzymes de la cholostase et du
prurit et retarde l’évolution défavorable.
Une transplantation
hépatique a transformé le pronostic des formes évoluées de la cirrhose
biliaire primitive. Parmi les modèles pronostiques récemment proposés pour
déterminer le moment optimal de la greffe, celui de Mayo clinic semble le
meilleur. Il fait intervenir des paramètres simples cliniques ou biologiques
(âge, bilirubinémie, albuminémie, PT, score d’oedème).
III. LA CHOLANGITE SCLEROSANTE PRIMITIVE
La cholangite sclérosante primitive est une
inflammation chronique des grosses voies biliaires intra ou extra-hépatiques,
évoluant vers la fibrose et provoquant une cirrhose avec hypertension portale
et insuffisance hépatique chronique. Elle se développe plus fréquemment chez l’homme, dans la tranche d’âge de 20 à
50 ans. Elle est associée dans plus de 60% des cas à la recto-colite
ulcéro-hémorragique. Une transformation néoplasique s’observe dans 10% des
cas.
La cholangite primitive doit être distinguée des
cholangites secondaires à des inflammations d’origine connue (lithiase
cholédocienne, sténose bénigne, ...).
Le diagnostic se fait par la cholangiographie RMN ou
rétrograde et la biopsie hépatique.
Le traitement est symptomatique comme dans toute cholostase chronique. La cholestyramine
peut apporter un soulagement du prurit et une antibiothérapie est indiquée en
présence d’une complication infectieuse. Comme traitement de fond, l’acide ursodésoxycholique a fait ses
preuves. Dans les formes extra-hépatiques compliquées de sténoses un traitement endoscopique par prothèse
est justifié. Dans les formes intra-hépatiques s’accompagnant d’angiocholite
chronique, d’ictère et/ou d’hypertension portale, on proposera une transplantation hépatique.
CHAPITRE VIII :
PATHOLOGIES HEPATIQUES METABOLIQUES
ET GENETIQUES
I. L’HEMOCHROMATOSE
L’hémochromatose est une maladie caractérisée par
une surcharge en fer.
Elle existe sous deux formes :
· L’hémochromatose primitive d’origine génétique transmise suivant un caractère autosomal récessif. La maladie ne se manifeste que chez une partie des homozygotes. Les hétérozygotes sont indemnes. La surcharge ferrique localisée dans les hépatocytes est due à une exagération de l’absorption intestinale de fer dont le mécanisme est mal connu. La mutation C282Y située sur un gène (dénommé HFE), sur le bras court du chromosome 6 à proximité des gènes HLA de classe I a été découverte récemment.
·
L’hémochromatose
secondaire au sein de laquelle on range les surcharges liées à des
transfusions sanguines répétées ainsi que les hémolyses et les dysérythropoïèse
(thalassémie). Une surcharge modérée peut être observées dans la cirrhose
particulièrement lorsqu’elle est d’origine alcoolique. Le fer est localisé dans
les cellules de Kupffer.
Le tableau classique de l’hémochromatose débute vers 40 ans chez les hommes et à la
ménopause chez les femmes.
L’asthénie et des arthropathies sont souvent les premières manifestations de
l’affection. Des douleurs de l’hypocondre droit associées à une hépatomégalie peuvent être observées au
début de la maladie. Une mélanodermie,
une atteinte cardiaque (troubles du
rythme, insuffisance cardiaque), un diabète, une atteinte gonadique et une
déminéralisation osseuse sont le reflet d’une atteinte avancée.
Les tests biologiques hépatiques sont normaux ou
montrent des altérations mineures dans les formes débutantes. Il existe
toujours une saturation en fer élevée.
La recherche de la mutation C282Y à l’état homozygote confirme le diagnostic d’hémochromatose génétique. Une biopsie peut être faite pour doser le fer tissulaire et évaluer les lésions histologiques, en particulier la fibrose.
La mise en évidence d’une hémochromatose génétique
chez un patient doit conduire à la réalisation d’une enquête familiale
(ascendants, collatéraux et descendants) par la recherche de signes biologiques
de surcharge en fer ainsi que celle de la mutation du gène HFE.
Le traitement comporte des saignées hebdomadaires de 500 ml. Ce traitement a d’autant plus de
chances d’être efficace qu’il est insaturé précocement. L’évolution du taux de
ferritine est un témoin fidèle de son efficacité.
En l’absence de traitement, l’évolution se fait vers la cirrhose et ses complications dont
l’hépatome.
II. LA DEFICIENCE HEREDITAIRE EN ALPHA-1 ANTITRYPSINE
Cette affection rare d’origine génétique peut être responsable chez les patients adultes homozygotes d’une hépatite chronique cirrhogène. Les manifestations pulmonaires peuvent dominer le tableau clinique. Le diagnostic est posé par le dosage de l’a1 antitrypsine et le typage phénotypique.
L’a1 antitrypsine est synthétisée par le foie et dans une moindre mesure par les monocytes et les macrophages, et elle migre dans la région de a1-globulines à l’électrophorèse des protéines sériques. Sa fonction principale est d’inhiber l’élastase leucocytaires.
Les taux normaux sont de 150-350
mg/dl, et augmentent en cas d’inflammation, de grossesse, ou lors de la prise
de contraceptifs oraux. Le diagnostic d’un déficit héréditaire hépatique en a1-antitrypsine
peut être supposé par la découverte d’une quasi absence de bande en a1-globulines
à l’électrophorèse, mais est confirmé par un dosage. L’a1-antitrypsine a un
polymorphisme génétique : 90% des caucasiens sont homozygotes pour l’allèle M,
les autres allèles étant F, S, Z et nul. La présence de l’allèle Z,
particulièrement dans les formes homozygotes, est associée à la maladie. Un
phénotypage par une technique de focalisation isoélectrique (séparation des
protéines selon leur charge) permet sa mise en évidence.
III. LA MALADIE DE WILSON
L’affection est caractérisée par l’accumulation de
cuivre dans les tissus, essentiellement le foie, le système nerveux central et
les yeux (anneau de Kayser-Fleischer). L’accumulation de cuivre est secondaire
à une élimination biliaire insuffisante.
Chez l’adulte jeune, l’affection se présente sur le plan hépatique comme une hépatite chronique active ou une hépatite fulminante. La biologie montre une réduction du taux sérique de céruloplasmine ; le cuivre sérique et la cuprurie de 24 heures sont élevés. Le dosage du cuivre hépatique sur prélèvement biopsique confirme le diagnostic.
Le dosage de la céruloplasmine sérique, la protéine
transporteuse du cuivre, est utile dans le diagnostic d’une maladie de Wilson.
Elle n’est pas impliquée directement dans ce trouble autosomique récessif de
stockage du cuivre, mais des concentrations basses (< 20 mg/dl) sont
retrouvées dans environ 90% des sujets homozygotes et 10% des hétérozygotes. Ce
dosage doit être accompagné de celui du cuivre sérique. En effet, la céruloplasmine
est une « acute phase protein » positive, mais sont taux augmente
aussi sous l’action des oestrogènes ou d’autres molécules comme la phénytoïne,
ce qui limite son interprétation si elle est dosée de manière isolée.
Les mesures diététiques (peu efficaces vu l’ubiquité
du cuivre) consistent à éviter l’absorption d’aliments riches en cuivre, tels
que le chocolat, les champignons, les coquillages, le foie et les arachides. La
drogue de choix est la D-pénicillamine (Kélatin*) qui augmente l’élimination
urinaire de cuivre ou le stocke dans les tissus sous forme non toxique. Le
traitement doit être poursuivi durant vie entière. Une interruption de ce
traitement peut s’accompagner de poussées évolutives ou fulminantes de
l’affection. La transplantation hépatique (qui corrige le trouble métabolique)
doit être réservée aux hépatites fulminantes, aux cas d'intolérance
médicamenteuse, d'absence de réponse thérapeutique et aux cirrhoses
décompensées.
IV. TROUBLES GENETIQUES DU METABOLISME DE LA BILIRUBINE
1. La maladie de GILBERT
C’est la cause bénigne la
plus fréquente d’hyperbilirubinémie non
conjuguée. Elle atteint 5 à 8% de la population et est transmise de manière
autosomique récessive. Le diagnostic est évoqué durant l’adolescence ou chez
l’adulte jeune. La plupart des patients sont asymptomatiques ou se plaignent de symptômes non spécifiques
(asthénie, nausées, ...).
L’ictère se
développe lors de périodes de jeûne, de stress, de fatigue, de prise d’alcool,
ou d’affection intercurrente. Le taux de bilirubine atteint en général 3 mg/dl,
mais peut -être supérieure. Les autres tests hépatiques sont normaux. Il est
nécessaire de rassurer le patient et de lui expliquer que la maladie est
bénigne et ne nécessite aucun traitement.
Les seules techniques réellement
diagnostiques sont des techniques chromatographiques permettant de déterminer
la quantité relative de bilirubine non conjuguée et de dérivés monoconjugués
dans le sérum, mais elles ne peuvent pas être réalisées en routine clinique.
2. Maladie de DUBIN-JOHNSON
Il s’agit d’un trouble
héréditaire rare (autosomique récessif) du transport canaliculaire de la
bilirubine conjuguée. La plupart des patients sont asymptomatiques et le taux
de bilirubine est compris entre 2 et 20 mg/dl. Il est important d’en faire le
diagnostic afin d’éviter toute investigation inutile. Les tests hépatiques sont
normaux. L’examen histologique du foie montre un pigment brun dans les
hépatocytes. Cette maladie est bénigne et il n’y a aucune sanction
thérapeutique.
3. Maladie de ROTOR
Elle représente aussi un trouble de la liaison conjuguée à la ligandine. C’est une maladie bénigne et il n’y a aucun traitement
CHAPITRE IX :
PATHOLOGIES HEPATIQUES VASCULAIRES
I. LE FOIE CARDIAQUE
En présence de processus
aigus de décompensation cardiaque
et en particulier d’états de choc, de troubles sévères du rythme, de coeur
pulmonaire aigu ou de cardiomyopathie aiguë, la réduction brutale du débit
sanguin est responsable de lésions hépatiques comportant la nécrose
centro-lobulaire. Leur gravité dépend de la durée de la réduction du débit
cardiaque. Les manifestations cliniques sont caractérisées par un ictère
généralement modéré. On relève une augmentation importante des transaminases et
une altération du temps de prothrombine. Une insuffisance hépato-cellulaire
mortelle peut être observée.
Dans la décompensation
cardiaque chronique, la péricardite constrictive, l’insuffisance
tricuspidienne, l’ictère peut être important ; il est d’origine
multifactorielle. Le diagnostic repose sur l’échographie cardiaque,
l’écho-Doppler des veines sus hépatiques et la biopsie hépatique. La tension de
la capsule hépatique provoquée par l’hépatomégalie brutale rend compte de la
sensibilité du foie à la palpation. La présence d’un reflux hépato-jugulaire
confirme le diagnostic. En présence d’une pression veineuse élevée, et
notamment en cas de péricardite constrictive, de l’ascite peut être observée et
être éventuellement de nature exsudative. Ce n’est que lorsqu’une cirrhose,
éventualité rarement observée, se développe (en présence de lésions
tricuspidiennes ou de péricardite constrictive) que des signes d’hypertension
portale se manifestent.
II. LE SYNDROME DE BUDD-CHIARI
Il s’agit d’une occlusion des veines sus-hépatiques dont les étiologies peuvent être nombreuses : cf. hypertension portale.
Dans les formes aiguës, on observe des douleurs abdominales, des vomissements, une hépatomégalie, du subictère et de l’ascite. Le décès peut survenir rapidement à la suite d’une faillite parenchymateuse. Les formes chroniques, plus fréquentes, provoquent des douleurs, de l’ascite et une hépatomégalie. L’ictère est rare. Les altérations enzymatiques sont généralement peu importantes. L’ascite peut être exsudative.
Les procédés suivants permettent de poser le diagnostic :
· écho-Doppler : l’absence de flux dans la veine sus-hépatique associée à une hépatomégalie plaide en faveur de cette affection ;
· résonance magnétique nucléaire : cette méthode est intéressante pour l’exploration non invasive des vaisseaux ;
· biopsie hépatique : une congestion avec hémorragie centro-lobulaire est l’image habituellement observée ;
·
venographie par voie fémorale ou jugulaire.
III. LA MALADIE VEINO-OCCLUSIVE
Elle est caractérisée par un
oedème et une fibrose, voire une occlusion des petites veines sus-hépatiques
(centro-lobulaires et sublobulaires). Les produits toxiques (alcaloïdes de la
pyrrolybdène contenu dans les plantes) ou des médicaments (azathioprine,
uréthane, dacarbazine) représentent l’essentiel de l’étiologie.
IV. LA PELIOSE HEPATIQUE
L’administration de contraceptifs oraux, de stéroïdes, peut être associée à des dilatations focales des sinusoïdes. Dans la péliose, on observe des cavités remplies de sang dont le diamètre peut varier de quelques mm à plusieurs cm.
Une hépatomégalie douloureuse associée à une augmentation des enzymes hépatiques peut être observée. L’arrêt de médicaments responsables provoque souvent une régression des lésions.
CHAPITRE X :
LES CIRRHOSES
I. ANATOMOPATHOLOGIE
Les cirrhoses se manifestent par une induration et par une perte de
l’architecture de l’organe dont l’aspect macroscopique devient nodulaire. Elles sont la conséquence
d’une destruction chronique progressive du parenchyme hépatique accompagnée
d’un fibrose. A l’examen histologique, les nodules, qui sont en remaniement
constant suite à la persistance des phénomènes hépatitiques et à la
régénération cellulaire compensatrice sont de taille variable suivant
l’étiologie et l’évolution. La classification des cirrhoses est difficile.
Classiquement, on utilise comme critère de distinction la taille des nodules.
Dans la cirrhose
micro-nodulaire ou cirrhose de Laënnec, le foie présente un aspect
« clouté » provoqué par la formation de petits nodules de la taille
d’un acinus hépatique. Le bord du foie devient dur et tranchant. Ces nodules
sont séparés par des septa fibreux minces, englobant à la fois des territoires
portaux et centro-lobulaires.
Dans la cirrhose
macronodulaire les nodules sont de taille irrégulière ; certains ne
dépassent pas le mm; tandis que d’autres ont des tailles de plusieurs cm.
L’aspect macroscopique du foie est extrêmement irrégulier, le bord tranchant
est fortement découpé. A l’examen microscopique, l’aspect des nodules est
variable ; certains sont composés de nids de cellules ne présentant aucune
organisation acinaire tandis que d’autres englobent des acini intacts.
Dans la cirrhose
de forme mixte on observe une combinaison de la cirrhose micro et
macronodulaire.
On admet classiquement que la cirrhose de Laënnec
est provoquée par l’éthylisme, tandis que la cirrhose macronodulaire est
habituellement la conséquence d’une hépatite virale. La réalité est cependant
moins simple ; en effet, une cirrhose éthylique, qui au départ est
micro-nodulaire, peut après de nombreuses années, suite à des remaniements
successifs du parenchyme, prendre un aspect macronodulaire.
A l’heure actuelle, la classification morphologique
est supplantée par une classification plus précise basée sur l’étiologie.
II. ETIOLOGIE ET CLASSIFICATION
Les causes les plus fréquentes
de cirrhose sont l’alcoolisme et les hépatites B et C. De multiples autres
étiologies sont possibles.
|
ETIOLOGIE DES CIRRHOSES |
|
|
|
Causes |
|
A.
PARENCHYMATEUSES Stéato-cirrhoses Virales Auto-immunes Médicamenteuses Métaboliques et génétiques Vasculaires Cryptogéniques B. CHOLOSTATIQUES Cirrhose biliaire
primitive Cirrhose biliaire
secondaire Cholangite sclérosante Médicamenteuses Mucoviscidose |
Alcool, Obésité,
Médicaments (Amiodarone) C, B,
B delta Trouble immunitaire INH, Dantrolène,
Méthyldopa Hémochromatose Maladie de Wilson Déficit en a1 antitrypsine Foie cardiaque Syndrome de Budd-Chiari Maladie veino-occlusive Cause indéterminée Trouble génétique et immunitaire Sténose biliaire chronique Trouble génétique et immunitaire Chlorpromazine, Ajmaline Trouble génétique |
III. MANIFESTATIONS CLINIQUES ET DIAGNOSTIC
A. STADE COMPENSE
Ce stade représente la
période la plus longue dans l’histoire naturelle de la cirrhose. Cliniquement
latent, il peut être une découverte au cours d’un examen de routine. Des
symptômes peu spécifiques peuvent être présents : asthénie, gêne de l’hypocondre droit. Dans certains cas, les
manifestations d’une hypertension portale (hématémèse
ou ascite) inaugureront une cirrhose préalablement asymptomatique.
L’examen peut montrer des angiomes stellaires, de l’érythème palmaire et une hépatomégalie avec
ou sans splénomégalie. Le diagnostic repose essentiellement sur les
éléments apportés par les aspects morphologiques précisés par l’imagerie
(échographie + Doppler) et une biopsie hépatique
éventuellement couplée à une mesure du gradient veineux hépatique. La
présence de varices oesophagiennes sera recherchée par endoscopie.
Les tests biologiques peuvent être strictement
normaux. Les plus sensibles et les plus fréquemment altérés au stade compensé
sont les suivants :
·
Le test de l’aminopyrine C14. La déméthylation de
l’aminopyrine C14 (prise par voie orale) par les microsomes hépatiques
(cytochrome P450) aboutit à l’élimination du C14 dans l’haleine où il est
mesurable. En cas de maladie hépatique, et notamment dans la cirrhose,
l’élimination du C14 est diminuée. Les valeurs normales vont de 4,2 à 9,3% en
deux heures.
·
Une
augmentation du taux d’acides biliaires et une diminution du taux de la
cholinestérase.
B. STADE DECOMPENSE
Les signes cliniques comportent, en général, la
présence d’un ictère, d’une ascite, d’une encéphalopathie et d’une hypertension
portale. Les autres complications (voir insuffisance hépatique chronique)
sont cardio-vasculaires et pulmonaires, endocriniennes, hématologiques,
cancérologiques (hépatocarcinome), certaines sont dues à la sensibilité aux
infections et aux médicaments.
Les examens biologiques montrent des taux plus ou
moins élevés de bilirubine, ainsi qu’une diminution du taux d’albumine et du
temps de prothrombine associée à une augmentation des gammas globulines. Le
taux de transaminases est fréquemment élevé. Le rapport AST/ALT est supérieur à
l’unité. Des manifestations d’hypersplénisme sont fréquentes.
L’imagerie peut montrer un foie hétérogène, aux
contours parfois bosselés, et des signes d’hypertension portale.
Le diagnostic est confirmé par la biopsie par voie
transveineuse.
IV. TRAITEMENT
Le régime doit être bien équilibré et comporter 60 g
de protéines/jour et 30 à 40 kcal/kg/jour.
La cirrhose alcoolique se traite par la suppression de l’alcool, les cirrhoses
médicamenteuses par l’arrêt du
médicament causal. L’interféron est peu utile pour la cirrhose
post-hépatique B et pour la cirrhose post-hépatique C compensées. La lamivudine est indiquée en cas de
cirrhose décompensée avec multiplication virale.
Le traitement des complications a été envisagé
antérieurement.
L’indication d’une transplantation hépatique dans la cirrhose sera précisée
ultérieurement.
V. PRONOSTIC
L’évolution de la cirrhose
est variable, dépendant en partie de sa cause. On considère que la présence
d’une décompensation avec complication de type ictère, ascite, hémorragie
digestive, infection bactérienne, encéphalopathie, sont des facteurs de mauvais
pronostic.
Dans le cas particulier de la cirrhose alcoolique, la poursuite de l’éthylisme ou l’apparition d’une hépatite alcoolique sont des facteurs d’aggravation.
Le pronostic de la cirrhose peut être estimé plus
objectivement de trois manières :
· Le test respiratoire à l’aminopyrine .
· Des tests hépatiques endogènes : acides biliaires et
cholinestérase.
· La classification de Child-Pugh.
|
EVALUATION DE GRAVITE D’UNE CIRRHOSE |
|||
|
Le score de Child-Pugh fait appel à critères
cliniques et biologiques. Pour chacun de 5 critères, une valeur de 1 à 3 est
octroyée suivant la sévérité du critère, ce qui permet d’établir un score
variant de 5 à 15. |
|||
|
Valeur du critère ® |
1 |
2 |
3 |
|
Encéphalopathie Ascite Bilirubinémie Albumine PTT |
0 0 <
2 mg/dl >
3,5 g/dl > 60% |
Modérée Modérée 2-3 mg/dl 2,8 - 3,5 g/dl 40 - 60% |
sévère sévère >
3 mg/dl <
2,8 g/dl <
40% |
|
CHILD-PUGH
A : score 5 à 6 CHILD-PUGH
B : score 7 à 9 CHILD-PUGH
C : score 10 à 15 |
|||
Les survies à 1 an sont approximativement les suivantes : 90% pour les stades A, 70% pour les stades B et 45% pour les stades C.
CHAPITRE XI :
ABCES, KYSTES ET TUMEURS HEPATIQUES
Ces lésions ne provoquent pas d’altération
fonctionnelle hépatique mais se manifestant par des symptômes liés à la présence d’une masse.
I. LES ABCES HEPATIQUES
A. LES ABCES A PYOGENES
1. Etiologie
Les abcès pyogènes du foie peuvent résulter des
affections suivantes :
· un
infection biliaire (30%) ;
· une
dispersion par voie sanguine via le système porte, d’infections abdominales (diverticulite,
appendicite), néoplasie (10%) ;
· une
septicémie généralisée avec envahissement du foie par l’intermédiaire de
l’artère hépatique ;
· une
extension directe à partir d’un foyer infectieux voisin (vésicule biliaire,
poumon).
Leur origine reste imprécise dans environ 50% des
cas. Les abcès hépatiques, de taille variable, peuvent être solitaires ou
multiples.
2. Manifestations cliniques et diagnostic
Le début peut être brusque ou insidieux. Le patient
présente des signes d’infection
(fièvre souvent oscillante, asthénie, anorexie) et d’une manière inconstante,
des douleurs de l’hypocondre droit,
parfois irradiées à l’épaule, souvent augmentées à l’inspiration. L’ictère est,
par contre, rare et tardif. L’examen révèle une hépatomégalie, une douleur à
la percussion de la base de la cage thoracique droite et des signes pleuropulmonaires de la base droite.
Les tests hépatiques restent le plus souvent
normaux. Il existe une hyperleucocytose et une élévation éventuelle des
phosphatases alcalines. La radiographie à blanc révèle parfois une surélévation
de la coupole diaphragmatique droite qui peut être déformée. Le diaphragme peut
être immobile. Un discret épanchement pleural droit n’est pas exceptionnel. L’échographie et la tomodensitométrie montrent la présence dans le foie d’une masse à
contenu liquidien finement hétérogène à contours irréguliers et flous. La paroi
devient hyperdense après injection de produit de contraste.
3. Traitement
Le traitement comporte, en général, une
antibiothérapie spécifique et, éventuellement, un drainage percutané de la
cavité abcédée.
B. LES ABCES AMIBIENS
1. Etiologie
Les amibes proviennent d’un foyer ulcéré de la paroi
intestinale. Ces ulcérations sont surtout observées dans le caecum ; cependant
elles peuvent également atteindre le côlon, le sigmoïde et le rectum.
L’Entamoeba histolytica atteint le foie par voie
portale. Les lésions parenchymateuses hépatiques sont secondaires à l’action
d’enzymes protéolytiques amibiens. Des abcès solitaires ou plus rarement multiples
en sont la conséquence. L’abcès hépatique amibien a, en général, le volume
d’une orange et siège, le plus souvent dans la partie antérieure du lobe droit. Son centre contient un pus chocolaté. Initialement, il n’y a
pas de paroi individualisée, celle-ci se forme ultérieurement.
L’abcès hépatique amibien peut compliquer une
amibiase aiguë ou ancienne. Le temps de latence entre le développement de
l’abcès et les lésions intestinales responsables de la dysenterie amibienne
peut être très long (jusqu’à 30 ans).
2. Manifestations cliniques et diagnostic
Les signes
cliniques, biologiques et radiologiques sont les mêmes que dans les abcès non
amibiens. Le début de la symptomatologie est habituellement progressif,
bien que des manifestations brutales aient été signalées. Les complications
principales sont la ruptures de l’abcès soit dans la cavité abdominale, soit
dans la cavité pleurale et la formation d’abcès cérébraux par voie sanguine.
La positivité des réactions sérologiques obtenues par immunofluorescence,
immunodiffusion ou hémagglutination représente l’élément le plus spécifique en
faveur de l’origine amibienne d’un abcès hépatique. La recherche des amibes
dans les selles est positive dans les amibiases aiguës. La mise en évidence
d’amibes dans le pus de l’abcès est très rare.
Lorsque la suspicion épidémiologique est grande, il
est conseillé d’entreprendre le traitement avant d’obtenir le résultat de ces
examens qui requièrent un long délai de réponse.
3. Traitement
Le traitement de choix de
l’abcès hépatique amibien est le metronidazole
(Flagyl*) administré à la dose de 750 mg, trois fois par jour durant 10 jours.
En vue d’éviter les récidives on conseille de stériliser le tube digestif par
un amoebicide de contact (Gabroral* 4/j – 10 jours). L’efficacité du traitement
sera évaluée par échographie : la lacune doit disparaître en 1 à 4 mois. Une
récidive est observée dans environ 50% des cas. En cas d’échec ou de récidive,
la vidange ou le drainage par ponction percutanée sous contrôle endoscopique
est la méthode de choix.
II. LES KYSTES HYDATIQUES
L’échinocoque est un parasite dont l’hôte définitif est le chien, l’hôte intermédiaire est le mouton, le boeuf et éventuellement l’homme. L’homme se contamine en ingérant des oeufs présents dans les selles du chien, en général, par léchage des mains ou caresse du pelage. Les oeufs dont la coque a été libérée par l’HCI se transforment en larves et celles-ci atteignent le foie par voie porte et y forment des kystes. Dans 20% des cas, les kystes se forment dans les poumons, 10% siègent ailleurs (rate, cerveau).
Le lobe droit du foie est atteint préférentiellement (85% des cas). Le kyste hydatique est limité par une enveloppe formée, du dehors en dedans, par :
· une membrane conjonctive adventice développée par l’hôte (périkyste) ;
·
une cuticule hyaline ;
·
une membrane proligère mince produisant les vésicules
filles.
1. Manifestations cliniques
Les symptômes subjectifs du kyste non compliqué sont
ou absents ou peu spécifiques (anorexie, diarrhée, pesanteur épigastrique ou de
la fosse iliaque droite). Parfois, apparaissent du prurit ou des poussées
récidivantes d’urticaire, secondaires à une sensibilisation de l’hôte au
liquide du kyste. Le plus souvent, c’est l’apparition d’une hépatomégalie chez un sujet ayant vécu
dans une région où l’échinococcose est
endémique qui fait suspecter le diagnostic. Il n’y a, en général, ni
cholostase, ni hypertension portale. La rupture dans le péritoine provoque des
signes de prurit et parfois même un choc anaphylactique. La rupture dans les
voies biliaires se manifeste par des signes
biliaires : angiocholite, ictère par rétention, surinfection de la cavité
kystique. Le kyste peut également se rompre dans les organes voisins,
poumons, péricarde, côlon, etc.
2. Diagnostic
L’échographie
et la tomodensitométrie révèlent la
présente d’une collection liquidienne.
La présence de calcifications de la paroi et de vésicules filles ou de sable
hydatique permet-tent d’affirmer la nature hydatique du kyste, mais ces signes
manquent dans 10% des cas.
Les tests hépatiques sont en général normaux. Une
éosinophilie est parfois présente.
La présence d’immunoglobulines
anti-échinocoque est quasiment constante. Les IgM disparaissent rapidement
après la destruction du parasite tandis que les IgG persistent pendant des
années.
La ponction est formellement contre-indiquée.
3. Traitement
Le traitement est chirurgical et il est réalisé en
deux étapes :
a. Destruction des scolex
Il est, en principe, indispensable de détruire les
vésicules filles avant toute ouverture du kyste. Ces vésicules contiennent en
effet des scolex susceptibles de se greffer sur le péritoine provoquant une
hydratidose péritonéale secondaire. La stérilisation du kyste est
traditionnellement obtenue par injection intra-kystique de solutions
hypertoniques. Le champ opératoire sera protégé par des compresses imbibées du
même sérum. L’efficacité de cette méthode est sujette à caution et il est
actuellement recommandé de traiter le malade par de l’albendazole
(anti-helmintique) durant plusieurs semaines avant et/ou après l’intervention.
b. Elimination du kyste
La méthode la plus couramment utilisée consiste en
une résection de la partie du kyste saillant à la surface du foie (dôme
saillant) ainsi que de la membrane proligère. Les communications vers les voies
biliaires seront recherchées et suturées. La cavité résiduelle sera drainée et
éventuellement bourrée par de l’épiploon. Une méthode plus agressive
(périkystectomie) comportant la résection non seulement du kyste mais aussi du
périkyste peut également être utilisée. De réalisation plus délicate, elle
permet d’obtenir des cicatrisations plus rapides. L’hépatectomie n’a que des
indications exceptionnelles.
III. LES TUMEURS HEPATIQUES
Ne seront reprises dans ce chapitre que les tumeurs
les plus fréquentes.
A. LES KYSTES NON PARASITAIRES
1. Kystes simples
Ce sont des lésions
congénitales très fréquentes (3% des individus). Ils sont formés à partir de
l’épithélium biliaire mais ne communiquent pas avec les voies biliaires. Leur
taille va de quelques mm à plusieurs cm. Ils peuvent grandie, mais la
croissance est lente.
Ces kystes sont asymptomatiques sauf s’ils
atteignent une grande taille ce qui est très rare.
L’échographie suffit en général au diagnostic. Elle montre la présence d’une masse ronde anéchogène à parois très fines. La tomodensitométrie confirme la nature liquidienne de la lésion et son caractère avasculaire.
Les kystes biliaires de petit volume ne nécessitent
ni surveillance, ni traitement.
2. Maladie polykystique
La polykystose hépato-rénale est une maladie
héréditaire rare. Les kystes rénaux précèdent les kystes hépatiques et
provoquent une hypertension et une insuffisance rénale. Chez les malades
survivant à l’affection rénale (transplantés), des kystes hépatiques multiples
peuvent se développer. Leurs seuls symptômes sont dus à un effet de masse. Le
traitement comporte des hépatectomies partielles et des fenestrations des
kystes restants.
B. L’HEMANGIOME DU FOIE
Il s’agit d’une tumeur vasculaire, de type
caverneux, probablement congénitale. Sa fréquence est d’environ 4%. La
croissance de la tumeur est, en général, très lente.
De petite taille, il est en général asymptomatique et est découvert
fortuitement au cours d’une laparotomie, d’une laparoscopie, d’un examen échographique
ou tomographique. Plus volumineux, il se manifeste sous la forme d’une tumeur
palpable, molle et parfois accompagnée d’un souffle. Les hémangiomes volumineux
peuvent se rompre spontanément ou à la suite d’un traumatisme. De la
thrombopénie ou de l’afibrinogénie peuvent survenir à la suite de coagulation à
l’intérieur de la tumeur.
A l’échographie
on trouve une masse pleine, hyperéchogène. En tomodensitométrie la masse est hypodense sans contraste, et après
injection de produit de contraste elle s’opacifie lentement de façon
centripète. Des images caractéristiques sont obtenues en résonance magnétique
nucléaire. La réalisation d’une biopsie est classiquement contre-indiquée mais
ses dangers ont été surestimés.
Un traitement
(hépatectomie) ne sera conseillé que pour les très exceptionnelles tumeurs
volumineuses et symptomatiques.
C. L’ADENOME HEPATIQUE
Il s’agit d’une tumeur bénigne, généralement unique,
souvent encapsulée. Elle est formée de travées plus ou moins régulières,
d’hépatocytes normaux ou quasi normaux. En général de petite taille, elle peut
exceptionnellement atteindre 20 cm.
Nettement plus fréquent chez la femme que chez
l’homme, son apparition est favorisée par la prise de contraceptifs oraux fortement dosés et par les stéroïdes androgènes
ou anabolisants.
Il peut se compliquer d’hémorragies intratumorales ou péritonéales.
Sa transformation
en carcinome est exceptionnelle dans les adénomes secondaires à la prise de
contraceptifs, plus fréquente après prise d’anabolisants.
Les lésions, en général asymptomatiques, sont le
plus souvent découvertes fortuitement au cours d’un examen échographique (masse iso ou hypoéchogène) ou tomodensitométrique (masse pleine vascularisée).
Un diagnostic de certitude est souvent difficile à
établir sur des biopsies et la
confusion avec un adéno-carcinome est possible.
L’arrêt des
contraceptifs
ou des anabolisants est recommandé et la grossesse est contre-indiquée. Une
résection chirurgicale doit être envisagée sauf s’il existe un risque à la
pratiquer.
D. L’HYPERPLASIE NODULAIRE FOCALE
Sa fréquence est beaucoup plus grande que celle de
l’adénome. L’hyperplasie nodulaire
focale est caractérisée par la présence d’une ou plus rarement de plusieurs tumeurs plus ou moins volumineuses, dont le
centre est occupé par une étoile fibreuse qui envoie des prolongements vers
la périphérie. L’examen microscopique montre que la tumeur est faite de nodules
d’hépatocytes séparés par des bandes de fibrose contenant des néocanaux
biliaires. L’hyperplasie nodulaire focale n’est pas une néoplasie mais
représente plus probablement une malformation. Elle affecte les deux sexes mais
est plus fréquente chez la femme.
L’hyperplasie nodulaire focale est en général asymptomatique. Comme l’adénome, elle
peut se compliquer d’hémorragies,
mais leur fréquence est nettement plus basse que dans l’adénome. La survenue de
ces hémorragies serait, selon certains auteurs, favorisée par la prise de
contraceptifs oraux. A la tomodensitométrie,
deux particularités orientent le diagnostic : l’étoile fibreuse centrale et
l’opacification précoce et homogène après injection du produit de contraste. La
scintigraphie peut montrer une
lésion isocaptante, ce qui n’est pas le cas pour l’adénome.
L’abstention ou l’arrêt des contraceptifs est
conseillée. Un traitement chirurgical n’est indiqué qu’en cas de complication
ou de doute diagnostique.
E. LES TUMEURS MALIGNES PRIMITIVES
1. Anatomopathologie
L’hépatocarcinome est la tumeur maligne du foie la plus fréquente. On distingue
plusieurs variétés : localisées sous forme d’une masse unique de taille
variable, multinoduclaire, diffuse avec présence de tumeurs multiples. Du point
de vue microscopique, l’hépatocarcinome est formé de travées cellulaires
pluristratifiées dont les éléments ressemblent aux hépatocytes par
l’architecture générale, mais en diffèrent par la taille plus grande et le
polymorphisme cellulaire. L’hépatocarcinome a tendance à envahir les branches
de la veine porte et plus rarement les veines sus-hépatiques.
Le
cholangiocarcinome dérive de l’épithélium des voies biliaires ; il est donc un
adénocarcinome plus classique. Il est beaucoup plus rare que l’hépatocarcinome.
Il peut être associé à la cholangite sclérosante.
Une forme rare de cancer primitif
est le carcinome fibro-lamellaire.
Il affecte les deux sexes principalement entre 10 et 35 ans, indépendamment des
facteurs étiologiques habituels. L’évolution en est lente.
L’hépatoblastome est une tumeur très rare de type embryonnaire rencontrée chez
le jeune enfant.
2. Epidémiologie et étiologie
Très fréquent en Afrique tropicale,
l’hépatocarcinome est rare en Europe. Il existe une prédominance très nette
pour le sexe masculin.
L’étiologie est dominée par la notion de cirrhose préexistante qui est retrouvée
dans plus de 80% des cas. Les taux d’hépatome sont nettement plus élevés par
les cirrhoses d’origine virale (20-40% dans l’hépatite B) qu’éthylique (5-15%).
Les virus B ou C n’agissent cependant pas comme oncogène, mais indirectement
par les phénomènes de régénération qu’ils entraînent. Plus discuté est le rôle
de certaines toxines alimentaires provenant du métabolisme de champignons
contaminant les céréales (aflatoxine).
3. Manifestations cliniques
Le diagnostic d’hépatocarcinome sera évoqué dans
deux circonstances principales :
·
la
découverte par échographie d’une
tumeur hépatique solide. Il peut s’agir d’une découverte de hasard ou d’un
examen réalisé à titre systématique chez un malade atteint d’une affection
susceptible de se compliquer d’un adénocarcinome hépatique (cirrhose) ;
·
l’apparition
chez un cirrhotique connu d’une douleur de l’hypocondre droit, d’une
hépatomégalie grossissant rapidement, d’une ascite réfractaire devenant
hémorragique, d’un ictère ou d’une fièvre inexpliquée.
4. Diagnostic
L’exploration fonctionnelle
hépatique met fréquemment en évidence des signes de cholostase anictérique (augmentation des phosphatases alcalines, et
des gamma GT) si le parenchyme hépatique est normal. Chez les cirrhotiques les
anomalies habituelles de la cirrhose seront observées.
Une augmentation du taux de l’a-foetoprotéine sera observée dans 60% des cas. L’importance de l’augmentation dépend
de la taille de la tumeur. Une augmentation du taux d’a-foetoprotéine n’est cependant pas spécifique
de l’hépatome malin. Elle peut être observée dans d’autres tumeurs malignes ou
au cours d’affections bénignes (hépatite virale, femmes enceintes).
Des syndromes
para-néoplasiques peuvent apparaître : polyglobulie, hypoglycémie et
hypercalcémie.
L’échographie,
la tomodensitométrie, la résonance magnétique nucléaire et l’artériographie révèlent la présence
d’images suggestives (masse pleine, vascularisée).
Une biopsie
(sous contrôle échographique ou laparoscopique) sera réalisée sauf si le
diagnostic est certain à l’imagerie.
5. Traitement
Lorsque la lésion est localisée, mesure moins de 5
cm et n’envahit pas les vaisseaux, une résection (hépatectomie) peut être proposée s’il n’y a pas de cirrhose ou si
la cirrhose est du type Child A. Le taux de récidive à 3 ans chez ces malades
est d’environ 30% alors qu’il dépasse 70% lorsque la tumeur a plus de 5
cm. Une transplantation peut
aussi être proposée en cas de nodule de moins de 5 cm associée à une cirrhose
Child A.
Pour les lésions multiples ou en cas de cirrhose
Child B ou C, une transplantation hépatique
doit être envisagée. Le taux de récidive est de 10% lorsqu’il n’y a pas plus de
3 tumeurs et qu’elles ne dépassent pas 5 cm. Le taux de récidive dépasse 75%
dans les cas moins favorables et de ce fait les centres de transplantations
refusent en général ces malades.
Les autres alternatives de thérapeutiques sont
l’alcoolisation sous échographie et la chimio-embolisation (injection
intra-artérielle de lipiodol ultra fluide et de cisplatine).
F. LES TUMEURS MALIGNES SECONDAIRES
Les métastases néoplasiques peuvent atteindre le
foie soit par voie portale, soit par voie artérielle. Les origines les plus
fréquentes sont : les tumeurs digestives, pulmonaires ou mammaires.
Les métastases sont le plus souvent multiples. Elles
se manifestent par la présence dans le parenchyme hépatique de nodules de
taille variable, de forme arrondie ; leur nombre et leur taille peuvent être si
importants que le foie triple ou quadruple de volume.
Les symptômes sont analogues à ceux des tumeurs
malignes primitives. La présence d’un gros foie bosselé doit faire penser à la
présence de métastases. Le diagnostic peut être confirmé principalement par
échographie, tomodensitométrie et laparoscopie.
Une hépatectomie
partielle réglée ou à la demande doit être proposée en cas d’atteinte d’un seul
lobe ou en présence de métastases aisément réséquables dans les deux lobes,
sans récidive locale et sans autre localisation néoplasique de la tumeur
primitive. Cette éventualité est malheureusement assez rare. La ligature de l’artère hépatique a été
proposée. Elle est basée sur l’observation suivant laquelle les métastases
possèdent une vascularisation exclusivement artérielle. Quelques régressions
ont été obtenues mais il ne paraît pas que le pronostic ait été nettement
modifié. La chimiothérapie par voie
générale ou par voie locale (perfusion vasculaire hépatique) a également
provoqué des régressions temporaires, son efficacité dépendant de la nature de
la tumeur primitive.
La survie est très variable mais dépasse rarement 6 mois.
CHAPITRE XII :
LA CHIRURGIE HEPATIQUE
I. LES RESECTIONS HEPATIQUES
La chirurgie hépatique a considérablement progressé
au cours des 20 deniers années. La mortalité réduite et le contrôle de sa
morbidité a permis d'élargir ses indications pendant que certains traitements
médicaux, endoscopiques et radio-interventionnels supplantaient la chirurgie
proprement dite dans l'hypertension portale, certains cholangiocarcinomes du
hile, etc….
Comme l'ont montré les chapitres précédents, on distingue les segmentectomies, les hépatectomies et les dérivations biliaires.
Fig. 18 L'hépatectomie
est indiquée surtout pour les tumeurs primitives, les métastases uniques ou
isolées et les tumeurs bénignes. L'anatomie segmentaire fu foie doit être
revue pour comprendre les différentes techniques. Trois arborisations conditionnent les
limites de section: 1.
La vascularisation
artérielle 2.
L'arbre biliaire 3.
Le réseau confluent
sus-hépatique. Le cas illustré montre un hépatocarcinome
sur foie sain situé sur les segments II, III, IV; une lobectomie gauche est
insuffisante et une tri-segmentectomie laisserait un segment I probablement
mal vascularisé voire envahi. Il s'agit donc d'une hépatectomie gauche
vraie emportant 4 segments. Toutes les variantes peuvent être proposées
si l'on respecte le fait que chaque territoire épargné doit être irrigué et
drainé.

B DB CB AB
Fig.19
A. Hépatectomie droite
vraie
B. Hépatectomie droite
élargie au IV
C. Trisegmentectomie gauche
D. Hépatectomie gauche
II.
LES DERIVATIONS BILIAIRES
|
Fig.19 A. Hépatectomie droite
vraie B. Hépatectomie droite
élargie au IV C. Trisegmentectomie gauche
D. Hépatectomie gauche |
La
cholédoco-entérostomie et l'hépatico-entérostomie consistent à monter une anse
grêle sur la voie biliaire extra-hépatique ou intra-hépatique et à restaurer la
continuité jéjunale par un montage en Y, au pied de l'anse. Toutes deux
favorisent un reflux de la flore intestinale dans les voies biliaires qui peut
occasionner l'angiocholite ascensionnelle.
Après
l'intervention, la présence d'une aérobilie ( air dans les voies biliaires
intra-hépatiques ) démontre la fonctionnalité de l'anastomose. Le principe est
le même que l'intervention de Kasaï pour atrésie congénitale des voies
biliaires.
III. LA TRANSPLANTATION HEPATIQUE
Depuis 1981, date du début de l’utilisation de la
cyclosporine en clinique, le nombre annuel de transplantations hépatiques (TH)
s’est accru de façon exponentielle tant aux USA qu’en Europe. Malheureusement,
une pénurie d’organes se fait sentir et les programmes tournent au ralenti. De
ce fait, plus que jamais, se pose pour le clinicien la question de définir au
mieux les indications de la TH ainsi que le moment idéal pour inscrire le
patient sur une liste d’attente.
1. Indications en fonction d'un syndrome clinique
Un prurit féroce et réfractaire à tout traitement
médical (ex. : cirrhose biliaire), une raréfaction osseuse avec douleurs
osseuses (ex. : cirrhose biliaire), une encéphalopathie hépatique chronique
invalidante, une ascite réfractaire (éventuellement traitée dans un premier
temps par un shunt intra-hépatique par voie transjugulaire) peuvent constituer
des indications en soi.
2. Insuffisance hépatique globale terminale
a. Fulminante aiguë ou subaiguë
Parmi les facteurs pronostiques, retenons le facteur
V, le PTT, le degré d’encéphalopathie, le taux de bilirubine, le degré
d’acidose (intoxication au paracétamol).
b. Chronique
·
Cirrhoses parenchymateuses
Certains éléments cliniques (ascite réfractaire,
ictère, encéphalopathie réfractaire, hémorragies répétées sur varices,
infection répétée du liquide d’ascite) ou biologiques (diminution de
l’albuminémie, diminution du PTT, augmentation de la bilirubine) sont des
critères utiles. Des scores clinico-biologiques (tels que le score de
Child-Pugh) apportent plus d’objectivité.
Quelques points spécifiques doivent être relevés :
- Le problème de la
cirrhose alcoolique. Dans notre pays l’alcoolisme est la cause de la
cirrhose dans 60% des cas au moins. Cependant, 5 à 15% des cirrhoses
alcooliques sont éligibles en vue d’une greffe. On sélectionne les patients
susceptibles de rester abstinents après la TH (sobriété de plus de 6 mois avant
la greffe, reconnaissance de l’éthylisme par le patient et sa famille, volonté
de s’en sortir) et ne présentant pas de complications extra-hépatiques de leur
alcoolisme.
- Le problème de la
cirrhose B. En cas de multiplication virale (HBeAg +, DNA +) le greffon se
réinfecte par le virus B. On ne soumettra à la TH que les patients sans
multiplication virale (HBeAg - ; DNA -) ou infectés par le virus B delta. Une immunoprophylaxie
par immunoglobulines spécifiques sera toujours réalisée.
- Le problème de la
cirrhose C. Après TH, le virus C récidive systématiquement sur le greffon.
Cependant, les lésions induites sont peu sévères et d’évolution lente.
·
Cirrhoses biliaires
Les résultats de la TH pour cirrhose biliaire primitive ou cholangite sclérosante sont parmi les meilleurs (plus de 75% de survie à 3 ans) en raison de l’hypertension portale peu importante et de l’absence de défaillance hépatique grave. L’évolution de la bilirubine est un critère important pour décider du moment de la greffe.
3. Insuffisance hépatique sélective
C’est l’exemple de l’hyperoxalurie avec un déficit
sélectif dans le foie et des complications rénales (greffe combinée foie +
rein).
4. Tumeurs ou pseudotumeurs irrésécables
Ce groupe comprend les hépatomes sur cirrhose, les
cholangiocarcinomes du hile, les métastases de tumeurs endocrines, des tumeurs
hépatiques rares (ex. : hépatome fibro-lamellaire) ou des pseudo-tumeurs
parasitaires (ex. : échinococcose alvéolaire).
Le choix de la TH par rapport à la résection
chirurgicale ou des traitements alternatifs non chirurgicaux reste l’objet de
controverses.
5. Techniques chirurgicales
La technique la plus ancienne est la transplantation
orthotopique d’un foie de cadavre.
La rareté des donneurs a imposé le développement de
nouvelles techniques. Il est possible de réaliser une bipartition du foie ce
qui permet la réalisation de deux greffes à partir d’un seul donneur. Le lobe
gauche du foie peut être prélevé chez un donneur vivant apparenté.
Au cours d’hépatites fulminantes, il est possible de
transplanter en position orthotopique ou hétérotopique un foie réduit ou
partagé sans résection du foie du receveur, ce qui permet l’exérèse ultérieure du
greffon et l’arrêt de l’immunosuppression après avoir constaté la régénération
du foie natif.
|
INDICATIONS DE LA TRANSPLANTATION HEPATIQUE |
|
I. INDICATIONS EN FONCTION D’UN SYNDROME CLINIQUE · Prurit féroce ou réfractaire · Ascite réfractaire · Encéphalopathie réfractaire II.
INSUFFISANCE HEPATIQUE GLOBALE TERMINALE · Fulminante aiguë ou subaiguë · Chronique * Parenchymateuse Alcool, virus B, virus B
delta, virus C Hémochromatose, Maladie de
Wilson, déficit en a1 antitrypsine Hépatite autoimmune, cryptogénique * Biliaire Cirrhose biliaire
primitive Cholangite sclérosante Cirrhose biliaire
secondaire III.
INSUFFISANCE HEPATIQUE SELECTIVE Hyperoxalurie IV. TUMEURS
OU PSEUDO TUMEURS IRRESEQUABLES · Hépatocarcinome sur cirrhose · Tumeurs vasculaires · Echinococcose alvéolaire |
|
CONTRE-INDICATIONS DE LA TRANSPLANTATION HEPATIQUE |
|
Age : > 65 ans Mauvaise
compliance, mauvais entourage, atteinte extra-hépatique (cardiaque,
cérébrale, pulmonaire, neurologique) sévère et irréversible. Infection extra-hépatique non contrôlée Maladie HIV Néoplasie extra-hépatique Néoplasie intra-hépatique de grande taille et/ou invasion de gros axes veineux |