PLAN
I.principes
généraux et particularités de la
A. abord de l’enfant opéré et de sa famille
B. éléments de monitoring et de surveillance
péri-opératoire
C. rappels physiologiques cardio-respiratoires,
métaboliques et nutritionnels
D. AGE GESTATIONEL, POIDS, et PATHOLOGIES
ASSOCIEES
II. diagnostic
et prise en charge des malformations
B. abord pluridisciplinaire de la hernie
diaphragmatique
A. Signes de détresse respiratoire périnatale
B. Causes chirurgicales de détresse respiratoire
chez le nouveau-né et le nourrisson
3. Emphysème lombaire congénital
IV. la
pathologie abdominale aiguë du nouveau-né
A. entérocolite nécrosante du nouveau-né
B. iléus et péritonite méconiale
C. les atrésies duodénales et iléales
V. la
pathologie bilio-pancreatique
A. L'atrésie
des voies biliaires
B. La
dilatation congénitale des voies biliaires
D. Les
tumeurs et abcès hépatiques
G. La
pancréatite idiopathique
VI. les
troubles de La motricité digestive
C. le reflux gastro-oesophagien
VII. les
anomalies de la paroi abdominale
A. les hernies ombilicale et épigastrique
B. l’omphalocèle et le gastroschisis
VIII. les
anomalies périnéales
IX. les
douleurs abdominales aigues de l’enfant
B. les fistules ano - RECTALES
XI. les masses abdominales et thoraciques
XII. les
tumefactions cervico-faciales
E. les dysembryomes
pre-auriculaires
F. la ranule
J. Le tératome
XIII. TRAUMATOLOGIE
COURANTE
XIV. orthopédie
courante
A. les anomalies du pied - le pied bot
B. la luxation congénitale de hanche
C. la boiterie - hanche irritable - LCP -
épiphysiolyse
D. scoliose et attitudes scoliotiques

Avant
propos
La
chirurgie pédiatrique n’est ni la chirurgie de l’adulte adaptée à l’enfant ni
la pédiatrie avec un bistouri.
La
chirurgie pédiatrique représente la prise en charge diagnostique et
thérapeutique des pathologies pédiatriques (0 à 15 ans en Europe ou 18 ans -
USA) pouvant nécessiter une technologie ou une stratégie chirurgicale.
La
pathologie pédiatrique répondant à ce critère s’étend du méningiome au pied
bot, en passant par la brûlure, la fente labio-palatine et l’appendicite aiguë.
Elle
reproduit chez l’enfant l’éventail impressionnant des spécialités chirurgicales
de l’adulte.
L’objectif
de ce cours de 10 heures en 2è doctorat est donc forcément très
limité.
Il
espère fournir à l’étudiant en médecine les principes généraux et les
particularités de la chirurgie de l’enfant en rappelant les éléments
physiologiques, psychologiques, anatomiques qui ponctuent la pathologie
pédiatrique.
Il
donne les recettes "diagnostiques " des grands syndromes
(détresse respiratoire, vomissements, hémorragies, douleurs abdominales,
scoliose, masses cervicales...).
Il
aborde le rôle du chirurgien dans les malformations néonatales.
Il
expose les pathologies courantes (hernies...) et évoque sommairement les
affections plus rares (pathologies hépato-biliaires, tumeurs solides...).
Il
ne peut être exhaustif et se réfère donc aux ouvrages sérieux tels que :
Textbooks
An Aid to
Paediatrice Surgery. Mac Mahon; Churchill Livingstone : p. 267.
Essentials of
Pediatric Surgery. Marc Rowe; Mosby : p/ 899.
Neonatal Surgery.
Lister & Irving; Botter worths : p. 733.
Gastro-entérologie pédiatrique. Navarro & Schmitz; Flammarion : p. 526.
Pediatric
Surgery. Welch; Year Book Medical Publishers : p. 3200.
Techniques de Chirurgie Pédiatrique. Pellerin & Bertin; Masson : p. 596.
Embryology for Surgeons.
Skandalakis & Gray; Williams & Welkins : p. 1101.
Pédiatrie. Reiner
& Lobut. Med Si Mc Graw Hill : p. 417.
Journals
Journal of
Pediatric Surgery.
Pediatric Surgery
International.
European Journal
of Pediatric Surgery.
Chirurgie Pédiatrique.
I. principes généraux et
particularités de la
chirurgie
pédiatrique
A. Abord de
l’enfant opéré et de sa famille
Lorsque
médecin traitant, pédiatre ou chirurgien sont confrontés à la décision
opératoire chez un enfant, ils doivent prendre en considération l’angoisse
particulière des parents qui s’ajoute à l’incompréhension de l’enfant.
A
la prise de responsabilité du clinicien, s’ajoute donc la prise en charge de
l’inquiétude de l’enfant qui dépend essentiellement de l’âge et de la gestion
du contact parental. Celles-ci sont plus aisément gérées par le pédiatre, le
chirurgien pédiatrique et l'infirmière pédiatrique dont l’expérience facilite
la mise en confiance.
1. "Quelques recettes"
La
prise en charge anténatale
Elle
sera envisagée dans le cadre de la malformation congénitale. Elle est
pluridisciplinaire (intégration avec obstétricien, généticien,
radio-pédiatre...). Elle exige beaucoup de temps. L’investissement parental
émotionnel est énorme. Il faut éviter à tout prix la culpabilisation de
l’entourage familial vis-à-vis de l’anomalie génétique.
Depuis
de nombreuses années, cette prise en charge pluridisciplinaire est appliquée
dès le diagnostic de l’anomalie anténatale. Dans un premier temps, la priorité
est donnée à l'évaluation de la malformation, de son traitement et des
séquelles possibles. Après, la décision de poursuite de la grossesse, une prise
en charge médico-chirurgicale et psychologique est proposée aux parents.
4
conditions anténatales peuvent être observées.
TABLEAU :
|
|
Anomalie
congénitale |
Traitement |
Séquelles |
|
1 |
Mineure (fente palatine, doigt surnuméraire) |
Parfaitement standardisé (palatoplastie, amputation) |
Quasi absentes |
|
2 |
Sévère (atrésie duodénale, C.I.V.) |
Connu mais complexe (duodeno-jéjonostomie, septoplastie) |
Mineures ou rares |
|
3 |
Très grave (hernie diaphragmatique, myéloméningocèle) |
Intensif, aléatoire ou expérimental
(ECMO, AREC...) |
Majeures, probables |
|
4 |
Lethale (encéphalocèle) |
Aucun |
Définitives |
Idéalement,
la mère est transférée vers un centre médico-chirurgical d’obstétrique et de
néonatalogie qui permet un accouchement dans les meilleurs conditions pour la
mère et l’enfant et une éventuelle intervention chirurgicale dans un bloc
opératoire unique.
On
préfère désormais transférer une mère porteuse d’un enfant porteur d’une
affection digestive, cardiaque ou autre vers un centre hospitalier capable de
gérer cette pathologie spécifique.
Ces
progrès plaident pour la promotion de structures hospitalières regroupant
généticiens, échographistes anténataux, néonatologues expérimentés en
prématurité sévère et chirurgiens pédiatriques spécialisés en pathologie
lourde.
Les
progrès dans l’identification des mutations ou de la transmission anténatale de
gènes et anomalies géniques responsables de pathologies nous invitent à évoquer
la certitude que la thérapie génique s’appliquera d’ici 5 à 10 ans aux
nouveau-nés et peut-être avant la naissance. La délétion du gène APC
(chromosome 18) - polypose familiale, et les mutations Ras ou Tp53 ob ( cancer
colorectal) conduisent aux premières tentatives de tranvections géniques.
Au centre néonatal
L’infirmier(e)
joue un rôle prédominant. Il (elle) cimente l’environnement du nouveau-né en
appréhendant les parents, surveillant les paramètres de l’enfant, favorisant
l’allaitement maternel (banque de lait).
Dès
que la mère quitte la maternité, elle rentre chez elle sans son enfant opéré.
Ceci constitue une frustration à laquelle elle peut être préparée par l’équipe
néonatale (notamment grâce au recours à une psychologue) si la diagnostic est
anténatal.
Si
celui ci est postnatal, les parents sont fréquemment désemparés. Lorsque la
malformation est apparente (fente labio-palatine, méromélie...) ou qu’il existe
un handicap même temporaire (feeding tube, colostomie, cathéter central,
cystocath...), le "qu’en-dira-t-on" prend fréquemment le dessus. Il
faut donc armer les jeunes parents afin qu’ils soutiennent questions, remarques
et regards de leur entourage.
S’ils
sont correctement informés de la pathologie et des plans, le retour en famille est généralement aisé. Lorsque le suivi
médical sera long, incertain ou coûteux, l’équipe chirurgicale informera
progressivement les parents (atrésie de l’oesophage, malformation périnéale
complexe, anomalie cardiaque grave, vertébrale...).
En consultation
La
conversation s’adresse à la "cellule familiale". Mère, père, enfant
(parfois grand-parents) sont un interlocuteur. Il faut éviter à tout prix les
"apartés".
On
évitera l’effet "blouse blanche". A cet effet, il faut rappeler que
le port de vêtements blancs n’est aucunement démontré comme un handicap dans la
relation enfant - médecin mais par contre, l’attitude générale et
l’environnement dans lequel se font ces consultations sont par contre
prédominants.
On
réduira l’attente, on diminuera le bruit, on favorisera la personnalisation des
soins par des équipes stables et par la connaissance du prénom de l’enfant.
2. Le rôle de l’âge
Avant
3 mois, le contact est essentiellement visuel; la relation médecin - patient et
certainement chirurgien - patient se fait souvent par l’intermédiaire de la
mère ou de l’infirmière. Attirons l'attention des jeunes médecins sur
l'existence d'une personnalité et de perceptions considérables de la part des
nourrissons vis-à-vis de l'entourage (médical).
De
3 à 18 mois, approche calme, voix posée, gestes doux
surtout au moment de l’examen; l’enfant est rarement méfiant et plutôt
collaborant.
De
18 mois à 4 ans, le contact oral est installé. Il existe
une compréhension partielle des événements. Les réactions individuelles de
l’enfant apparaissent. Les parents jouent un rôle majeur dans la confiance
envers le pédiatre ou le chirurgien.
w Le pire : "on a pas dit à la petite qu’on venait chez vous pour une dilatation
anale, sinon c’était le drame". Elle s’en est rendu compte dans le
hall de l’hôpital !
w L’idéal : "L’enfant a été
parfaitement informé de ce qui l’attend à la consultation. Un petit cadeau
symbolique du chirurgien efface rapidement les éventuels pleurs d’un enfant
ayant souffert".
Après
5 ans, l’enfant raisonne vite, exige des informations
de plus en plus précises. Le chirurgien s’adresse de plus en plus à l’enfant
sans personne interposée. Ce n’est plus : "déshabillez-le, madame et restez près de l’enfant tant que je l’examine
mais plutôt : viens Valérie, montre-moi ton ventre, nous allons enlever les
fils".
La
rentrée scolaire joue un rôle prédominant puisque l’enfant déteste être
différent des autres. On essaiera de lui donner une autonomie maximale
(continence anale et urinaire, appareillage orthopédique, corrections
esthétiques...).
3. L’hospitalisation
La
plupart des institutions favorisent l’hospitalisation de type mère - enfant.
Celle-ci
n’est bien entendu pas possible dans les unités de réanimation dans les centres
de néonatalogie.
Après
une hospitalisation prolongée au centre néonatal, on recourt au "rooming
in". Celui-ci permet de rapprocher mère et enfant pendant quelques jours
afin de simuler une sortie de l’hôpital après un accouchement. Ce "rooming
in" permet à la mère de se refamiliariser avec un contact de 24 heures
auprès de son enfant. Depuis 1980, la chirurgie pédiatrique ambulatoire s'est
développée. Elle s'adresse aux gestes chirurgicaux simples et concerne les
enfants parfaitement entourés.
B. Eléments de
monitoring et de surveillance péri-opératoire
Si
la réanimation et la chirurgie néonatales sont sous l’unique responsabilité des
médecins hospitaliers spécialistes dans les centres néonataux (type N), dès les
premiers stages, l’externe et l’interne peuvent être confrontés à la surveillance
d’un nouveau-né ou d’un prématuré malade. L’ignorance totale des notions de
base (rythme cardiaque, rythme respiratoire, hémodynamique, homéothermie, ...)
conduit quelquefois l’étudiant à une attitude inappropriée voire dangereuse.
On
se souvient d’une jeune interne demandant à la mère d’un nouveau-né en
nutrition parentérale totale si c’était bien son lait qui coulait dans la
perfusion !... et du médecin de garde récemment promu qui avait commandé 2
litres de sang total pour une intervention chirurgicale prévue chez un enfant
de 3 kg ! Si le ridicule ne tue jamais le futur médecin, une température
rectale à 35,5°C met un prématuré de 2 kg en situation critique.
Quelques
considérations néonatales médicales interfèrent avec la surveillance néonatale
et celle de l’enfant spécifiquement "chirurgical".
Parmi
celles-ci rappelons :
a) L’immaturité pulmonaire du prématuré
-
maladie à membranes hyalines
-
rôle du surfactant
b) La dysplasie bronchopulmonaire
-
ventilation artificielle
-
le pneumothorax sur hypoplasie ou hyperinsufflation pulmonaire
c) La toxicité de l’oxygène
-
fibroplasie rétrolentale
d) L’hémorragie intra-ventriculaire (50% des
poids < 1500 gr)
e) L'immunodépression
f) Les affections iatrogènes
Toutes
ces conditions affectent et compliquent la surveillance des nouveau-nés
prématurés et enfants chirurgicaux.
Enfin,
sur le plan nutritionnel, les réponses métaboliques postopératoires observées
chez l’adulte s’appliquent au nouveau-né et au prématuré.
La
réponse catabolique est proportionnelle à la gravité de la pathologie
sous-jacente et à la lourdeur de l’acte chirurgical ainsi qu’à l’infection
éventuelle.
Le
recours à des sources énergétiques alternatives pour répondre aux besoins
accrus prend un sens particulier lorsque l’on sait que la réserve lipidique est
d’autant plus faible que la prématurité est grande.
A
la lipolyse réduite se substitue la glycogénèse. Si la modulation des réponses
au stress est moins documentée chez le nouveau-né et a fortiori chez le
prématuré, les observations cliniques étayent de nombreuses hypothèses
logiques. Le monitoring péri-opératoire doit aussi considérer la labilité
thermique, le contrôle de la douleur postopératoire, les besoins
protéo-caloriques, l’altération de la réponse immunitaire à l’infestation
normale (contamination du tube digestif) ou à l’agression par un agent
pathogène spécifique.
1. Monitoring hydro-électrolytémique
En
clinique, il n’existe actuellement aucun moyen de prévoir, précisément pour un
patient donné, les besoins hydriques. Quatre notions guident le dialogue avec
les espaces hydriques du patient.
a) "L’ouverture
thérapeutique" commence par l’administration de 150 à 180 ml/kg/24h. Elle
est classiquement décrite comme le "starting volume".
b) La
réponse du patient se fait par l’adaptation de l’osmolarité et du volume
urinaire, une modification de l’osmolarité plasmatique et des modifications de
poids corporel.Ce dernier est difficile à évaluer chez le prématuré au
respirateur.
c) L’ajustement
doit tenir compte de l’examen clinique, des pressions artérielles et veineuses
(fermeture du canal artériel, trou de Botal,...) et de l’environnement
(couveuse, table Ohio, photothérapie, homéothermie,...).
d) Le
bilan hydrique :
![]()
apport hydrique idéal
= apport actuel - excès d’urine.
Les
besoins sodés de base sont, chez l’enfant chirurgical, de 2 mEq/kg/24h; ces
besoins passent à 3 mEq pour les enfants les plus critiques.
Les
besoins potassiques de base sont de 1 à 2 mEq/kg/24h. En cas d'hypokaliémie
(sténose du pylore, occlusion digestive...), on peut raisonnablement corriger
la kaliémie de + 1 mEq par l'administration de 1 mEq/kg de poids corporel.
2. Monitoring respiratoire
La
méthode de choix permettant le contrôle non invasif de la fonction ventilatoire
est la "pulse" oxymétrie.
La
mesure de la saturation de l’hémoglobine en oxygène (SpO2) est
facile, atraumatique, corrélée à la SaO2; elle se fait par un sensor
apposé en général à l’un des orteils et se base sur le principe de la
spectrophotométrie.
Les
progrès réalisés par rapport à la mesure intra-artérielle ou percutanée ont été
parallèles au développement de nouvelles technologiques en matière d’assistance
ventilatoire.
Parmi
celles-ci citons le contrôle de la ventilation par la mesure constante des
résistances ventilatoires (feed-back immédiat que n’avaient pas les
ventilateurs à pression ou volumes imposés), la ventilation à haute fréquence
et basse pression (HFV ou jet ventilation) et plus récemment l’assistance par
oxygénation à membrane extra-corporelle (ECMO ou AREC).
Si
cette dernière technologie n’a pas encore prouvé scientifiquement un apport
substantiel en terme de survie des enfants atteints de malformations
congénitales (pas d’étude randomisée prospective), elle devient une alternative
envisagée dans la pneumopathie à membranes hyalines, l’hypoplasie pulmonaire et
la hernie diaphragmatique.
3. Monitoring cardiaque
Sans
revenir sur les notions de base de l’adaptation cardio-circulatoire du foetus à
la vie extra-utérine, il est utile de citer les techniques de monitoring
spécifiques cardiaque et hémodynamique.
- Le
rythme cardiaque
Reste
l’indice clinique fiable car il change immédiatement en cas de stress ou
d’hypoxémie. Un rythme cardiaque de 120 à 160/min est normal chez l’enfant à
terme.
Il
est plus élevé (130 à 170/min) chez le prématuré.
La
bradycardie inférieure à 100 est un indice capital de souffrance néonatale en
période péri-opératoire.
- La
pression systolique
Sa
mesure la plus fiable se fait par cathétérisme artériel radial ou fémoral. Il
faut noter qu’il s’agit d’une technique invasive qui n’est réservée qu’à des
enfants à haut risque. En effet, la pathologie iatrogène artérielle causée par
les cathétérismes inappropriés reste significative.
- Pression
veineuse centrale
La
mesure de la pression veineuse centrale est facile lors de l’acquisition d’un
cathétérisme droit. Il faut préciser que la mesure doit être faite par
transduction et non pas par manométrie. En effet, la variation rapide de
l’ampliation thoracique et des pressions pleurales en cours de ventilation
artificielle rend l’interprétation de la pression veineuse centrale mesurée par
manométrie difficile.
- Pression
pulmonaire occlusive (wedge pressure)
Lors
de l’existence d’un cathéter pulmonaire, la mesure de pression occlusive permet
de témoigner de l’évolution des résistances pulmonaires. Celles-ci augmentent
dans les pneumopathies à membranes hyalines, et dans l’insuffisance cardiaque
gauche.
Le
recours au cathétérisme de Swang-Ganz est rare chez le nouveau-né et chez le
prématuré mais il peut néanmoins dans certaines circonstances permettre une
mesure des débits cardiaques lors des situations cardiologiques particulières.
- Saturation
en oxygène du sang mêlé
La
mesure de la saturation en oxygène du sang mêlé dans les vaisseaux pulmonaires
par cathétérisme pulmonaire est devenue depuis peu un excellent témoin de
l’évolution des pathologies à haut risque sur le plan cardio-vasculaire (hernie
diaphragmatique,...).
4.
Monitoring
thermique
L’homéothermie
déjà labile chez le nouveau-né est totalement insuffisante chez le prématuré.
La mesure constante de la température rectale ou oesophagienne est
indispensable dans la phase per et postopératoire.
A
côté de la mesure de la température, les mesures prophylactiques sont bien
entendu préférables. Citons par exemple le recours à l’emballage des membres
par du coton ou une couverture de survie. L’utilisation systématique d’un
bonnet de coton évite la déperdition thermique significative au niveau du cuir
chevelu.
Si
au centre néonatal on recourt à l’utilisation de couveuses et de tables
chauffantes, le quartier opératoire préfère le chauffage par lampe, le recours
au matelas et le contrôle de la température ambiante de la salle d’opération
(28° dès le début de l'intervention chirurgicale).
C. Rappels
physiologiques cardio-respiratoires, métaboliques et digestifs
De
la surface corporelle, à la maturation enzymatique en passant par
l’homéothermie, la plupart des aspects physiologiques diffèrent chez l’enfant
par rapport au patient adulte. Cette assertion est à fortiori prédominante chez
le nouveau-né ou le prématuré. Quelques particularités d’intérêt clinique
doivent être notées.
1. Anatomie
w Cardio-pulmonaire
L’hémodynamique
foetale est conforme à la circulation foetale et placentaire. Ceci sous-entend
la persistance du shunt gauche-droit, le flux artériel et veineux important des
vaisseaux ombilicaux (ligaments ronds).
Toute
pathologie néonatale grave entraînant une augmentation des pressions
pulmonaires fait persister ou réinverse les gradients de pression gauche-droit
et modifie leur support anatomique (canal artériel, communication inter
auriculaire...).
w Osseuse
Le
squelette du foetus et de l’enfant est en partie cartilagineux. Tous les os
longs et plats présentent une relative souplesse des corticales. Ceci modifie
la réponse de la structure osseuse et cartilagineuse aux contraintes
traumatiques. Même en dehors de troubles de l’ostéogenèse, les diaphyses
peuvent se rompre en "bois vert"
(exemple : radius décubitus).
La
traumatologie tient compte de ce phénomène puisqu’en cas de fracture d’un des
os, l’os intact (exemple cubitus) peut empêcher la réduction du segment
fracturé en jouant le rôle de hauban.
De
même, la rupture en bois vert d’une seule corticale peut rendre le réalignement
et la fixation difficiles.
La
fracture iatrogène peut être nécessaire dans certains traumatismes afin
d’obtenir une réduction axiale des deux os contigus.
Le
cartilage de conjugaison est le maître mot de la traumatologie pédiatrique
puisque son respect conditionne la croissance. Tout matériel d’ostéosynthèse a
son niveau doit être évité car il déclenche ou accélère son ossification
prématurée.
Autre principe de la traumatologie
pédiatrique, l’absence de point d’ossification, au jeune âge, rend les
glissements épiphysaires difficiles à diagnostiquer radiologiquement.
L’échographie
et la résonance magnétique nucléaire ont récemment facilité l’identification
des lésions épiphysaires (coudes, cheville...); mais l’échographie est un
examen "opérateur dépendant". Il faut donc s’assurer de l’expérience
pédiatrique du radiologue; quant à la résonance magnétique nucléaire (RMN),
elle reste encore limitée à certains grands centres hospitaliers et est souvent
maîtrisée par des neuro-radiologues peu motivés par la pathologie osseuse
pédiatrique.
La
connaissance des délais d’apparition des points d’ossification et de leur
fusion s’avère, en revanche, fort utile pour traiter les malformations
ostéo-articulaires graves (scoliose , méromélie....).
Par
exemple, le signe de Risser (ossification de la crête iliaque) permet de
connaître le stade de croissance de la colonne vertébrale. Il oriente donc la
chronologie thérapeutique des scolioses (traction, corset, fixateur externe,
intervention chirurgicale ouverte).
w La descente du testicule et la fermeture du
canal péritonéo-vaginal est tardive pendant la gestation. La naissance
prématurée, par l’augmentation de pression intra-abdominale, peut empêcher
cette fermeture. Le pédiatre et le chirurgien pédiatrique sont donc confrontés
à une anatomie inguinale particulière chez le prématuré et le nouveau-né.
La
persistance d’un canal péritonéo-vaginal perméable conduit à l’hydrocèle
communiquant. Sa fermeture (90% avant un an) interrompt l’alimentation de
l’hydrocèle qui se résorbe, la vaginale étant absorptive et non sécrétante chez
l’enfant (se référer au cours d'Urologie du Prof. Schulman).
2. Rappels embryologiques digestifs
Il
est bien nécessaire de revoir l’organogenèse (surtout tardive) pour comprendre
certaines malformations congénitales et souvent identifier et interpréter leur
présentation.
a) La
rotation de l'anse vitelline autour d’une axe sagittal représenté par les
vaisseaux ombilicaux, prolongement de l’axe mésentérique supérieur se fait
selon un sens trigonométrique (90°).
Il
s’en suit une seconde rotation de 180° qui amène dans un plan frontal (10è
semaine) le mésentère et le cadre colique.
Le
duodénum "frontalisé" encadre la fusion des ébauches pancréatiques
ventrales (Wirsung) et dorsales (Santorini).
La
rotation de 270° coïncide avec la réintégration des viscères dans la cavité
abdominale (malrotation intestinale).
b) L’histogenèse
conduit à l’apparition des villosités (8 à 10 semaines), des cellules à mucus
(9 à 10 semaines), des cellules à gastrine, sécrétine et cholécystokinine (12
semaines).
c) La
rupture de la membrane cloacale ouvre le canal anal mettant en continuité la
lumière intestinale avec la cavité amniotique.
Dès
la 18è semaine, le sphincter externe devient contractile et augmente alors la
pression du canal anal. Ce tonus met fin à la perméabilité du canal anal
(atrésie anale).
3. Aspects métaboliques
Au
niveau de la physiologie digestive proprement dite, on rappellera le rôle
d’alcalinisation par le liquide amniotique dégluti pendant la période foetale.
Le
pH gastrique de 7 à la naissance passe à 4 après une heure sauf dans le cas de
la prématurité. La sécrétion acide est de 0,01 mEq/kg x heure.
A
un an, la sécrétion gastrique atteint 0.20 à 0.40 mEq/kg x heure, 0,2-0,4
mEq/kg.h.
On
note une gastrinémie élevée à la naissance opposée à une résistance ou une
saturation des récepteurs des cellules pariétales. Ce mécanisme peut intervenir
dans la sténose du pylore.
Au
niveau de la physiopathologie biliaire, on peut noter l’immaturité de nombreuses
chaînes enzymatiques hépatiques et la tendance à la cholestase du nouveau-né
notamment lorsque celui-ci n’est pas nourri par voie entérale. L’apparition de sludge vésiculaire entraîne quelquefois
une lithiase vésiculaire et une cholécystite du nourrisson.
Le
rôle du liquide amniotique est celui du liquide extra-cellulaire. On rappellera
l’apparition de l’urine foetale vers 4 mois et la déglutition de 500 ml de
liquide amniotique soit un apport de 50% du liquide extra-cellulaire corporel.
Toute atrésie oesophagienne duodénale ou jéjunale conduit donc à un hydramnios.
D’autres
chapitres tels les particularités immunologiques de l’enfant sont
éventuellement à revoir pour comprendre le rôle joué par la barrière
immunitaire intestinale notamment lorsque celle-ci est rompue en cas
d’entérocolite nécrosante.
Les
interférences avec la nutrition parentérale de l’enfant au niveau de la
maturité de la barrière immunitaire intestinale sont certaines. Le début de
l’alimentation entérale par l’apport d’allergène alimentaire déclenche une
série de processus indispensables à l’immunisation de l’enfant.
4.
Aspects nutritionnels
Si
tout porte à croire que l’apport calorique est inutile pendant les premières
heures et probablement pendant les deux premiers jours de vie (phase
catabolique initiale), très rapidement l’enfant et certainement le prématuré a
besoin d’un support nutritionnel complet adapté à ces besoins particuliers.
La
difficulté de recourir au tube digestif de l’enfant atteint de pathologie grave
peut nécessiter l’apport intraveineux de solution de nutrition.
L’impossibilité
de recours à la nutrition entérale est fréquent en cas de prématurité sévère,
de ventilation artificielle, d’iléus, d’obstruction digestive ou de sepsis
grave.
Le
contenu des solutions parentérales est en principe calculé selon un menu à la
carte et sous flux laminaire dans les centres de néonatalogie.
Certains
centres font appel à des solutions commercialisées en général peu adaptées au
besoin spécifique de chaque patient.
En
effet, le calcul des besoins en eau, ions, acides aminés, glucides et
éventuellement lipides est calculé très précisément par le néonatalogue en
fonction de chaque cas.
D. AGE
GESTATIONEL, POIDS et PATHOLOGIES
ASSOCIEES
Comme
il est largement démontré dans le chapitre consacré au nouveau-né normal, il
existe de multiples corrélations entre l’âge gestationel, le poids et les
problèmes médico-chirurgicaux rencontrés.
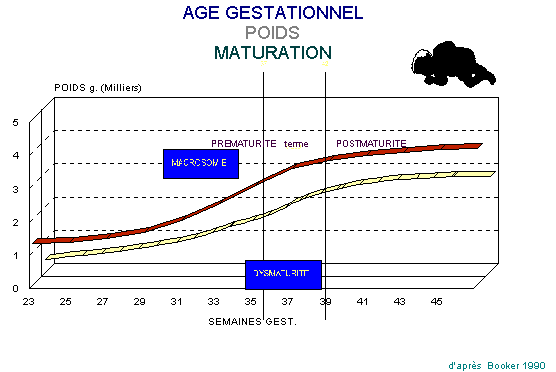
L’incidence
des malformations congénitales peut éminemment varier d’un pays à un autre,
surtout si les méthodes de détection et d’identification varient.
A
titre purement illustratif, les fréquences suivantes sont mentionnées dans les
registres américains:
TABLEAU /1000 naissances: extrapolation à la Belgique
10.000.000
habitants
120.000 naissances
Malformations cardiaques 10 1200
Malformations urinaires 3 360
Malformations digestives 3 360
Luxation de la hanche 2 240
Malformations des membres 2 240
Malformations neurologiques 1.5 180
Fentes labio-palatines 1.5 180
Pied bot 1 120
Syndrome de Down 1 120

Le
chirurgien pédiatrique est souvent confronté au problème des voies d’accès chez
l’enfant. Ce problème concerne bien entendu le pédiatre, le radiologue pédiatrique
et l’anesthésiste. Dans certaines institutions pédiatriques, un "IV
team" a été instauré. Celui-ci est responsable de la cartographie veineuse
périphérique et centrale de l’enfant. Il permet d’utiliser au maximum les
territoires veineux sans épuiser ceux-ci.
Ceci
est particulièrement important en néonatalogie et lors de l’administration
d’antibiotiques et d’agents chimiothérapiques à long terme.
a. Les veines épicrâniennes
Elles
sont fréquemment utilisées en routine par les pédiatres et néonatologues chez
les enfants en bas âge. L’absence de cuir chevelu développé et le caractère
translucide de la peau permet de reconnaître aisément les multiples veines
épicrâniennes. Si les aiguilles et cathéters qui sont installés doivent être de
diamètre réduit, le problème est essentiellement la fixation de la perfusion à
la tête de l’enfant.
Chez
les enfants qui ne sont pas immobilisés, une surveillance étroite est
nécessaire. Le déplacement de l’aiguille peut entraîner un écoulement
paraveineux et une nécrose accidentelle du cuir chevelu.
Les
épicrâniennes sont donc utilisées en général en urgence pour des perfusions
temporaires.
b. Veines périphériques des membres
L’utilisation
des veines périphériques du système radial et basilique ainsi que des veines
saphènes par les anesthésistes est fréquente. Il permet l’introduction de
cathéters de silastic et la perfusion pendant de nombreuses heures de solution
éventuellement irritante. Ces voies d’abord permettent rarement
l’administration de haut débit et notamment celle de dérivés sanguins. Ces
veines périphériques ont intérêt à être épargnées dans la mesure où les gestes
sous anesthésie peuvent être fréquents chez l’enfant porteur de malformations.
Ces
veines sont quelquefois dénudées chirurgicalement. La dénudation rend
quelquefois service chez les enfants de plus de 6 mois dont le tissu adipeux
est beaucoup plus développé. Les veines saphènes notamment sont devenues moins
palpables et leur dénudation facilite l’abord.
En
revanche, la dénudation veineuse ne prolonge pas la durée de vie de la
perfusion et entraîne un préjudice esthétique évident.
Une
autre technique consiste à introduire un cathéter central parfois périphérique.
Il s’agit des cathéters cutanéo-cave (nouille). Ces cathéters centraux par
accès direct périphérique sont guidés sur le principe du Seldinger. Ces
cathéters remarquablement évolués sur le plan technique sont peu traumatiques,
sont parfois le siège de thrombose. Leur positionnement doit être vérifié sous
amplificateur ou sous examen radiologique.
CARTE
VEINEUSE PERIPHERIQUE

c. Veines centrales
En
cas d’urgence chez le polytraumatisé ou lors de problèmes néonatals graves,
l’abord jugulaire interne, jugulaire externe ou sous clavier peut être utilisé.
La jugulaire interne est le premier choix pour l’introduction des cathéters
centraux par voie directe.
Le
cathétérisme de la veine jugulaire interne est fréquemment utilisé par les
anesthésistes pour l’obtention de gros débits perfusionnels.
Lors
de la nécessité d’administration prolongée de nutrition parentérale, il est
souhaitable de recourir à un cathéter tunnelisé. Celui-ci est constitué d’un
manchon de silastic introduit sous la peau.

Un
trajet de plusieurs centimètres sous cutanés réduit les contaminations
bactériennes classiques dans les cathéters centraux à long terme. La veine
jugulaire est dénudée et canulée par un cathéter de Broviac. Le tableau suivant
résume les avantages et les inconvénients.
TABLEAU
|
|
ACCES |
OPERATEUR |
DUREE |
COMPLICATIONS |
INDICATIONS |
|
EPICRANIENNE |
Facile |
Pédiatre |
HEURES |
labilité |
réhydratation |
|
PERIPH
PERCUTANEE |
Moyen |
Pédiatre ou
anesthésiste |
JOURS |
phlébites
et positionnel |
idem
+ médication |
|
PERIH DENUDEE |
Moyen |
Pédiatre ou
chirurgien |
JOURS |
phlébite,
nursing |
idem
+ chirurgie |
|
CUTANEO-CAVE |
Moyen |
Pédiatre |
SEMAINES |
thromboses |
n.parentérale brève |
|
CENTRAL
PERCUTANE |
Moyen |
Pédiatre,
chirurgien ou anesthésiste |
SEMAINES |
dangereux sepsis |
n.
parentérale longue |
|
BROVIAC |
Difficile |
Chirurgien |
MOIS |
thrombose (exige
narcose) |
n.
parentérale prolongée et chirurgie ou chimio |
II. diagnostic et prise
en charge des malformations in
utero
L’étiopathogénie
des malformations congénitales donne lieu à une littérature extrêmement
abondante qu’il est difficile de résumer dans ce cours. Si la grande majorité
(+ de 60%) des malformations congénitales reste d’origine inconnue, des progrès
constants sont faits dans l’identification du rôle génétique souvent
prépondérant.
Près
de 10% des anomalies congénitales ont un support génétique démontré. Près de 6%
sont constitués d’anomalies chromosomiques. Le rôle environnemental a été
démontré chez l’homme dans 5% des anomalies congénitales. Enfin, chez près de
20% une étiologie génétique et environnementale combinée semble la plus
plausible.
Un
bébé "anormal" naît sur la terre toutes les 30 secondes.
La
première confrontation lorsqu’un enfant naît porteur d’une anomalie a
généralement lieu si le déficit est apparent entre les parents et l’accoucheur
ou l’infirmière. Très rapidement, l’équipe néonatale médico-chirurgicale est
confrontée à l’abord psychologique du problème.
Trois questions
reviennent classiquement
:
-
est-ce la faute de l’un des parents ?
-
peut-on guérir ou corriger cette malformation ?
-
peut-on faire un autre enfant ?
Il
faut bien entendu éviter à tout prix une culpabilisation génétique des parents
et une enquête génétique doit être initiée. La deuxième question nécessite bien
entendu une culture générale et une connaissance des pronostics des affections
les plus fréquentes et elle constitue l’un des objets de ce cours. A la
troisième question, l’identification d’une transmission héréditaire ou d’une
mutation doit être faite. Elle conduit certainement à la surveillance
ultrasonique et génétique des grossesses ultérieures.
Parmi
les affections congénitales à composante héréditaire, on peut citer les
affections suivantes : fente labio-palatine, mucoviscidose, iléus méconial,
atrésie oesophagienne, luxation de la hanche, dysplasie acétabulaire, maladie
de Hirschsprung, hydrocéphalie, certaines méromélies, tératomes
sacro-coccygien...
Parmi
les facteurs exogènes responsables d’affections congénitales
(tératologie environnementale), on peut citer : la rubéole congénitale, le
diabète maternel, l’irradiation, l’hypothyroïdie par prise de substance
thyréoprive (cortisone, lugol), le tabagisme paternel et maternel, l’éthylisme
maternel et certains accidents traumatiques obstétricaux.
Le
diagnostic anténatal est avant tout basé sur la symptomatologie anténatale de
nombreuses affections.
Citons
par exemple :
|
Arthrogrypose |
Rareté des
mouvements foetaux |
|
Tumeur de Wilms |
Volume utérin
supra normal et image ultrasonique caractéristique |
|
Atrésie
digestive haute |
Hydramnios |
|
Agénésie rénale
bilatérale |
Oligoamnios |
|
Méromélie grave |
Diagnostic
ultrasonique |
D’autres
affections peuvent être suspectées par l’anamnèse familiale (mucoviscidose,
polypose familiale, certaines anomalies urinaires...). L’incidence des
anomalies des malformations touchant les nouveau-nés varie selon les organes et
les systèmes.
Sur
1000 naissances, l’incidence malformative globale est de 20 à 30.
On
note par ordre de fréquence décroissante : les malformations cardiaques (10),
les malformations digestives (3), les malformations urinaires (3), les
luxations de la hanche (2), les malformations des membres (2), les
malformations neurologiques (1.5), les malformations faciales (1.5). VOIR TABLEAU page 15
La
prise en charge d’une grossesse peut se résumer selon le TABLEAU ci-dessous
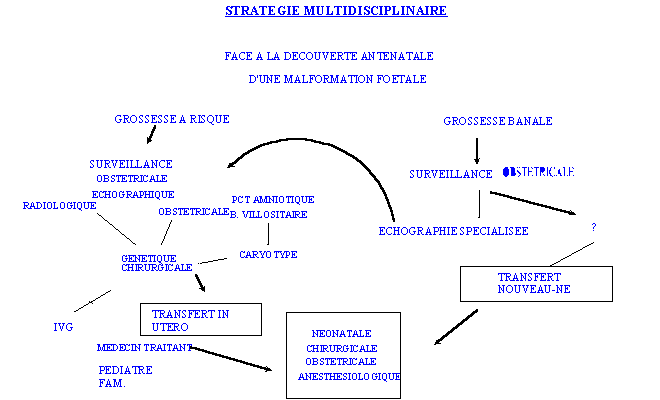
B. Abord
pluridisciplinaire de la hernie diaphragmatique
La
hernie diaphragmatique congénitale de Bochdalek est choisie comme anomalie
anténatale nécessitant une prise en charge pluridisciplinaire. Des aspects
physiologiques et cliniques sont particulièrement étendus et caractéristiques
de la prise en charge néonatale.
1. Définition
Parmi
les 3 pathologies aiguës du diaphragme - hernie, éventration et paralysie, la
hernie de Bochdalek constitue la forme la plus spécifique d’anomalies
diaphragmatiques. Le diaphragme présente embryologiquement un orifice naturel,
hiatal et deux sites de faiblesse congénitale possibles : l’orifice
postéro-latéral - hernie de Bochdalek - et l’orifice rétrosternale - hernie de
Morgani . La hernie postéro-latérale gauche de Bochdalek consiste en
interruption de la coupole diaphragmatique et à une protrusion intra-thoracique
des viscères abdominaux avec hypoplasie pulmonaire et déviation du bloc
cardio-pulmonaire. L’incidence est de 1 sur 3000 naissances. Il s’agit de
l’urgence néonatale la plus grave.
L’étiologie
consiste en un arrêt de croissance des coussins diaphragmatiques entre la 8è
et la 10è semaine de la gestation.
Avant
cette période, les cavités thoraciques et abdominales foetales communiquent
largement. Le septum transversum sépare les deux cavités progressivement
d’avant en arrière. Les brèches postéro-latérales doivent se fermer vers la 8è
semaine. L’arrêt de cette croissance et de la fusion des coussins entraîne le
passage aisé des organes abdominaux vers le thorax. Le développement alvéolaire
pulmonaire est donc compromis essentiellement du côté homolatéral. La
compression mécanique par les ébauches digestives en croissance continue
refoule progressivement le bloc cardiaque et peut induire l’hypoplasie
pulmonaire hétérolatérale. La présence d’une hypertension pulmonaire à la
naissance induit la persistance du shunt droit - gauche. La déglutition d’air
augmente le volume du tube digestif intra-thoracique. En l’absence de
traitement, l’évolution naturelle conduit au décès dans les heures qui suivent
la naissance.
2. Symptomatologie
Le
diagnostic anténatal est devenu pratiquement systématique et conduit à une
évaluation génétique complète incluant amniocentèse, biopsie villositaire et
caryotype.
Une
surveillance écho-cardiaque approfondie est proposée. Les symptômes, à la
naissance, sont centrés sur la dyspnée, le ventre plat, le thorax bombé de type
emphysémateux et une cyanose ne répondant pas à l’administration d’oxygène. La
ventilation au masque aggrave la situation respiratoire et cardiaque. Le bruit
de murmure vésiculaire est absent du coté atteint et des borborigmes
intestinaux peuvent être entendus dans les heures qui suivent au niveau
thoracique. Le diagnostic anténatal est fréquent. Le diagnostic clinique à la
naissance est simple et la radiographie de thorax confirme bien entendu la
protrusion diaphragmatique. Elle est beaucoup plus fréquente à gauche mais elle
peut atteindre la coupole droite ce qui entraîne une hernie et une anomalie
anatomique hépatique. La brièveté des veines sus hépatiques constitue du reste
un obstacle à une réduction aisée du foie intra-thoracique.
3. Prise en charge immédiate et
aspects physiopathologiques
Dans
la mesure du possible, l’enfant est gardé en ventilation spontanée avant une
intubation endo-trachéale. La sonde gastrique est placée rapidement et
raccordée à une aspiration douce intermittente.
Si
la ventilation traditionnelle au respirateur haute pression risque de provoquer
une rupture alvéolaire dans le poumon hypoplasique (pneumothorax), on lui
préfère une ventilation à basse pression et à haute fréquence de type "jet
ventilation". Le pronostic a été considérablement amélioré par le recours
à ce type de ventilation. Certaines équipes expérimentent la circulation
extra-corporelle par oxygénateur à membrane (ECMO ou AREC). L’hypoplasie
pulmonaire majeure constitue une bonne indication mais aucune étude randomisée
n’a actuellement démontré clairement les avantages. Alors que l’intervention
chirurgicale était considérée jadis comme urgente, elle est actuellement
différée de quelques heures voire de quelques jours afin d’obtenir une
stabilisation ventilatoire hémodynamique de l’enfant.
4. Traitement chirurgical
Les
enfants sont équipés d’une cathéter central de type Broviac afin de permettre
une nutrition parentérale. La pression intra-abdominale est monitorisée par
l’intermédiaire de la sonde gastrique. L’intervention chirurgicale consiste en
une laparotomie sous costale gauche ou sous costale droite éventuelle. La
réintégration des viscères permet le repositionnement dans la cavité
abdominale. Il est à noter qu’une malrotation partielle ou totale est associée
puisque les accolements n’ont pu se réaliser pendant la vie intra-utérine. En
cas de malrotation, l’appendicectomie est réalisée systématiquement.
La
brèche diaphragmatique congénitale peut être suturée soit per primam, soit par l’utilisation d’une greffe musculaire
pédiculée, soit par l’interposition d’un matériel synthétique de type Téflon ou
Goretex. Le matériel résorbable est bien entendu proscrit.
La
réintégration des organes dans la cavité abdominale et la fermeté du diaphragme
permettent de gagner partiellement au niveau de la ventilation pulmonaire;
celle-ci est essentiellement conditionnée par l’hypoplasie pulmonaire et non
par le volume thoracique disponible.
Par
contre, au niveau abdominal, une hyperpression intra-abdominale peut réduire le
retour veineux cave inférieur, la perfusion rénale voire la perfusion
mésentérique.
Le
chirurgien évite de dépasser une pression intra-abdominale de 20 cm d’eau en
phase aiguë et de 15 cm d’eau de manière continue.
Le
pronostic est essentiellement lié à l’hypoplasie pulmonaire et la récupération
d’une oxygénation correcte dans la phase postopératoire. Le drainage pleural
systématique permet de contrecarrer un éventuel pneumothorax. La persistance de
la circulation de type foetal avec cyanose et canal artériel constitue des
facteurs péjoratifs. Des drogues tonicardiaques et les agents surfactants sont
régulièrement évalués.
5. Pronostic
En
terme de santé publique, les chiffres historiques évoquent une mortalité
globale de près de 50%. Ce chiffre tend à se modifier d’une part par le biais
d’interruption volontaire de grossesse et d’autre part, par une meilleure prise
en charge multidisciplinaire de cette pathologie. On estime qu’une survie de
70% constitue un excellent indice de réussite.
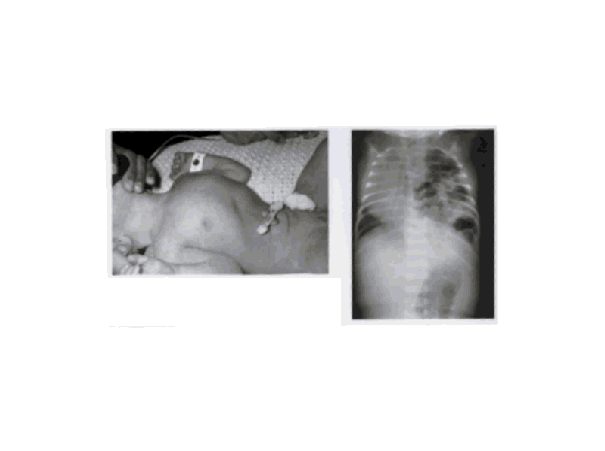
|
Hernie Diaphragmatique Notez
le thorax bombé, le ventre plat, L’enfant
est immédiatement intubé sans ventilation au masque. |
Volumineux
défect diaphragmatique,
Les anses grêles et le colon sont
intra-thoraciques ; shift médiastinal important et hypoplasie pulmonaire
bilatérale |
La
détresse respiratoire néonatale constitue une pathologie médico-chirurgicale de
gravité importante nécessitant une reconnaissance et une prise en charge
immédiate. Le score d’Apgar constitue le premier reflet de la qualité de la
ventilation puisqu’il fait appel à une mesure de la respiration, de la
fréquence cardiaque, de la réaction à l’aspiration buccale et de la coloration.
De
multiples particularités anatomiques et physiologiques expliquent l’instabilité
respiratoire du nouveau-né; citons : l’étroitesse des voies respiratoires
supérieures et la rapidité d’une obstruction mécanique; le développement
insuffisant de la musculature intercostale rendant le diaphragme essentiel dans
la ventilation. Toute compression abdominale des coupoles diaphragmatiques
réduit le volume courant et interfère avec l’oxygénation sanguine. La
concentration tissulaire d’oxygène des enfants et du nouveau-né en particulier
est nettement supérieure par rapport à l’adulte (+ 50%).
A. Signes de
détresse respiratoire périnatale
- Fréquence
respiratoire inférieure à 20 ou supérieure à 60 par minute;
- Battements
des ailes du nez;
- Tirage
intercostal diaphragmatique sous sternal ou gémissements;
- Acrocyanose
et cyanose distale;
- Convulsions;
- Coma.
Les
signes de retentissement cardio-vasculaire sont une tachycardie fixe, associée à
un choc et une oligurie.
Les
origines de la détresse respiratoire au cours des premières 24 heures sont
liées à :
a) étage
supérieur (atrésie des choanes);
b) thorax
(immaturité pulmonaire, pneumopathie à membrane hyaline, pneumothorax et
pneumomédiastin, fistule trachéo-oesophagienne et malformation cardiaque);
c) paroi
(hernie diaphragmatique, éventration diaphragmatique).
Après
36 heures, la détresse respiratoire évoque plutôt des diagnostics de
pathologies abdominales (obstruction digestive), de malformations cardiaques,
de mucoviscidose, d’emphysème lobaire ou de broncho-pneumonie)
La
cyanose dite calme fait penser à un trouble neurologique primaire ou une
cardiopathie. Quelles que soient les multiples causes de dyspnée, l’obstacle
sur les voies respiratoires supérieures doit systématiquement être recherché
par aspiration buccale naso-pharyngée.
Après
celles-ci, des causes chirurgicales de détresse respiratoire peuvent être
évoquées.
B. Causes
chirurgicales de détresse respiratoire chez le nouveau-né
et le nourrisson
Atrésie
des choanes, syndrome de Pierre-Robin, trachéomalacie, compression extrinsèque
(lymphangiome).
Fistule
trachéo-oesophagienne, emphysème lobaire congénital, pneumopyothorax, hernie
diaphragmatique, éventration diaphragmatique.
Elle
consiste en une imperforation congénitale des choanes qui peut être unilatérale
ou bilatérale. Le passage systématique de sonde nasale par l’infirmière au
moment de la naissance doit pouvoir exclure ce diagnostic.
L’atrésie
des choanes unilatérale passe parfois inaperçue et conduit rapidement à un
jetage nasal. Elle est bénigne et conduit à des troubles O.R.L. secondaires.
L’atrésie bilatérale conduit à une détresse respiratoire. Il faut signaler que
le jeune enfant ne respire pas spontanément par la bouche. L’absence de ce
réflexe conduit à une asphyxie. La cyanose et le tirage immédiat après
l’accouchement signent le diagnostic. L’introduction d’un tube de Mayo ou de
Guedel supprime immédiatement l’asphyxie. Dans un premier temps, l’alimentation
de l’enfant nécessite le recours à une tétine coupée. Très rapidement, la cure
chirurgicale transpalatine est proposée. Le pronostic est bon après chirurgie.
Le
syndrome de Pierre-Robin associe une hypoplasie mandibulaire et une fente palatine.
La rétrognatie
et l’implantation anormale de la langue conduisent à une obstruction
des voies aériennes supérieures. L’enfant doit être immédiatement positionné
sur le ventre avec une éventuelle traction linguale. L’enfant est nourri par
feeding tube gastrique pendant les premières semaines. Un appareillage
prothétique peut être installé temporairement si la détresse respiratoire est
grave. Dans les formes mineures, le pronostic est bon puisqu’une croissance
mandibulaire secondaire spontanée permet la correction de l’anomalie. Il
n’existe aucun déficit esthétique à long terme.
3. Emphysème lombaire congénital
Relativement
rare, cette anomalie est associée à une chondromalacie et à des atypies
vasculaires. Elle induit une hyperinflation brutale d’un lobe pulmonaire. La
dyspnée d’apparition progressive est associée à une cyanose. On note un murmure
vésiculaire abaissé, un tympanisme et un shift médiastinal.
Le
traitement est chirurgical et réalisé par thoracotomie. Il consiste à réséquer
les bulles emphysémateuses. L’évolution est incertaine en fonction de la
gravité de la chondromalacie et de l’étendue de l’affection.
L’ensemble
des formes d’atrésie de l’oesophage sera traité dans ce chapitre parce que
cette affection digestive est
associée à des complications respiratoires qui ponctuent le tableau clinique.
Pathogénie
Au
23è jour du développement embryonnaire, un diverticule ventral se
forme à la paroi antérieure du tractus digestif supérieur primitif. Deux
bourrelets latéraux se rejoignent et vont séparer l’oesophage de la trachée
vers le 35è jour. La séparation des deux structures peut donner lieu
à de multiples anomalies de développement dont l’atrésie de l’oesophage avec
fistule trachéo-oesophagienne basse.
L’anomalie
atteint le feuillet endodermique et peut prendre une forme polymalformative de
nature typiquement génétique (syndrome de Vater) ou plus ponctuelle touchant
éventuellement une mutation spécifique (chimère).
Symptomatologie
L’incidence
est de 1 sur 2000 naissances.
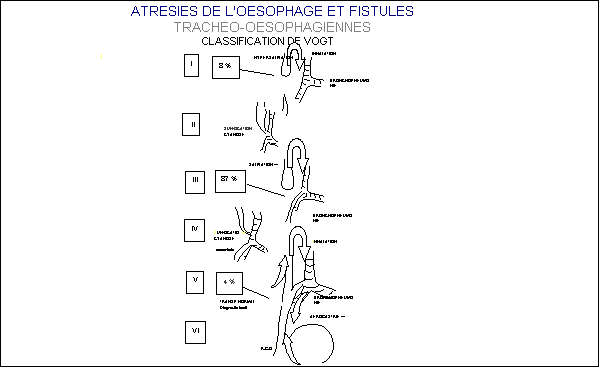
La
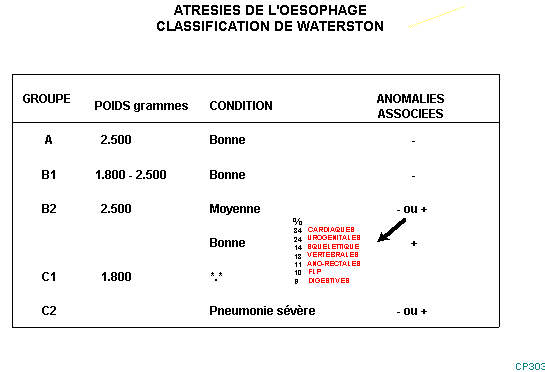
symptomatologie dépend du type d’atrésie. Le tableau indique 6 formes classiques
selon la classification de Vogt.
.
Le type 3 est le plus fréquent et consiste en une dilatation du circuit
oesophagien supérieur et une fistule trachéo-oesophagienne distale à hauteur de
la caréna. Le type 1 et le type 5 sont classiques mais moins fréquents. Les
types 2 et 4 sont exceptionnels.
Le
type de symptomatologie est explicable par anatomie de la lésion. Il faut
ajouter que l’hypersalivation induit fréquemment une inhalation même en
l’absence de fistule trachéo-oesophagienne.
La
symptomatologie du type 3 consiste en une salivation excessive avec toux et
inhalation Suffocation et cyanose secondaire
Un
ballonnement abdominal complique le tableau en l’absence de geste
thérapeutique.
L’anomalie
est quelquefois diagnostiquée ou suspectée en période anténatale (présence
d’hydramnios). L’anomalie associe le plus fréquemment la perforation anale.
Le
diagnostic du type 3 est classiquement réalisé par une radiographie de thorax
montrant une image aérique borgne visible en face des vertèbres cervicales. La
présence ou non d’air dans la cavité abdominale signe la présence d’une fistule
broncho-oesophagienne ou trachéo-oesophagienne distale.
La
forme la plus difficile à diagnostiquer est la fistule trachéo-oesophagienne
sans atrésie oesophagienne.
Elle
peut être diagnostiquée tout à fait secondairement par des affections
respiratoires récidivantes.
Attitude thérapeutique
L’enfant
est positionné en décubitus ventral et position anti-Trendelenbourg. Une sonde
d’aspiration dans le réservoir oesophagien évite la déglutition de salive.
L’enfant est perfusé et n’est ventilé qu’en cas de détresse respiratoire.
Le
traitement est chirurgical et consiste le plus fréquemment en une thoracotomie
droite postéro-latérale avec abord extra-pleural de l’oesophage. La fistule
trachéo-oesophagienne est refermée et la continuité oesophagienne est rétablie
en évitant la traction.
Le
recours à une nutrition parentérale ou à une gastrostomie peut être nécessaire.
Dans les formes les plus graves, une
oesophagostomie et une gastrostomie sont réalisées. Une reconstruction
oesophagienne par gastroplastie ou coloplastie est réalisée vers l’âge d’un an.
IV. LA PATHOLOGIE ABDOMINALE AIGUE DU NOUVEAU-NE
A. Entérocolite nécrosante du nouveau-né
Si l’entérocolite nécrosante (NEC) ou
entéropathie vasculaire est décrite depuis 1891 son intitulé précis date du
début des années 60. Affection mortelle et rare, cette pathologie a vu son
incidence considérablement augmenter dès le début des années 70. Les progrès
spectaculaires réalisés dans la réanimation des enfants de petit poids et de
grande prématurité ont entraîné l’éclosion de cette affection dans les pays
occidentaux. Après un pic dans les années 1980, l’incidence s’est stabilisée au
cours des dernières années. Un traitement médico-chirurgical approprié a
amélioré considérablement son pronostic.
Si l’affection est rare voire exceptionnelle
chez l’enfant de poids de naissance normal et en bonne santé elle atteint
jusqu’à 25% des enfants dont le poids est inférieur à
1200 g.
Sur le plan anatomo-pathologique, l’affection
consiste en une souffrance ischémique intestinale localisée ou extensive
conduisant à la nécrose et à la perforation en péritoine libre.
De la phase initiale oedematiée et hémorragique
à la nécrose installée, plusieurs stades histologiques peuvent être décrits
incluant des ulcérations aux muqueuses, l’exsudation fibrineuse et la
pneumatose intestinale consistant en bulles aériques présentes dans la
sous-muqueuse du grêle ou du colon. La prolifération bactérienne dans les
régions ulcérées est nette. Au niveau microscopique, on note un sludge et des
micro-thromboses. L’entérocolite nécrosante peut être associée à des atrésies
intestinales ou à une maladie de Hirschsprung à laquelle elle est alors
secondaire.
Pathogénie
De multiples facteurs peuvent déclencher
l’entérocolite. Le point-clé de cette étiopathogénie est certainement une chute
du débit vasculaire entraînant une souffrance cellulaire muqueuse et
sous-muqueuse, des hémorragies et une prolifération bactérienne sur un terrain
immunodéprimés. D’une oxygénation insuffisante pour raison respiratoire jusqu’à
la dilatation intraluminale sur Hirschsprung ou iléus méconial tout incident qui
concourt à interrompre ou réduire le transport en oxygène vers la muqueuse
intestinale peut être impliqué.(voir tableau)
Actuellement de nouvelles hypothèses
passionnantes sillonnent les esprits.
Si l'enfant prématuré est mis en contact avec
les allergènes apportés par la nutrition ou la contamination bactérienne
orodigestive, on peut imaginer l'activation brutale des lymphocytes présents
dans la muqueuse et la sous-muqueuse intestinale. La libération des cytokines
et la brutalité de la réaction inflammatoire pourrait induire une réaction
endothéliale brutale altérant l'oxygénation tissulaire. La prédominance colique
serait alors attribuable à la précarité vasculaire plutôt qu'à la quantité
absolue de lymphocytes prédominant, eux, dans la paroi intestinale grêle.
Ces réflexions demandent prudence et recherche.
Tableau
clinique
Les signes cliniques sont strictement calqués
sur les facteurs déclenchants et sur le stade idiopathogénique où se trouve le
nouveau-né.
Afin de bien comprendre l’évolution clinique, la
description des symptômes et signes radiologiques est empruntée à Belle (1973).
Stade I
(suspicion)
L’histoire clinique révèle un stress périnatal.
L’instabilité thermique, l’asthénie ou l’irritabilité, l’apparition d’apnées et
de bradycardies sont les signes généraux.
Sur le plan digestif le ballonnement abdominal,
une sensibilité à la palpation profonde et des hémorragies occultes dans les
selles peuvent être observées. La radiographie d’abdomen à blanc démontre une
hétérogénéité de la répartition aérique et une distension localisée de
certaines anses grêles. Un tableau de type pseudo-occlusif peut être décrit
radiologiquement.
La biologie est peu contributive si ce n’est une
chute éventuelle de la leucocytose et des plaquettes.
Stade II (confirmé)
On note une aggravation de l’état général
précédemment décrit. L’hémorragie intestinale peut être macroscopique. La
distension abdominale douloureuse ne fait plus aucun doute. L’alimentation
orale devient impossible, les clichés radiographiques démontrent la présence
d’un iléus sévère, d’une augmentation d’épaisseur de la paroi intestinale avec
oedèmes interstitiels et une pneumatose intestinale ou portale. Ces signes
peuvent également être objectivés par un échographiste pédiatrique confirmé.
Des répercussions cardio-vasculaires sévères peuvent être observées. La
biologie peut être peu contributive.
Stade III (avancé)
Détérioration clinique et répercussion
hémodynamique. Apparition d’un choc septique éventuellement sévère et
aggravation de la symptomatologie abdominale. L’échographie ou la radiographie
d’abdomen montre un pneumopéritoine. La nécrose intestinale est éventuellement
installée et la perforation exprimée cliniquement. La péritonite généralisée
peut s’ensuivre ainsi que le décès.
La
prise en charge thérapeutique
Dès le stade I, l’alimentation orale doit être
interrompue.
Ceci est d’autant plus vrai que le début de
l’alimentation entérale chez un prématuré sévère peut être le facteur
déclenchant. L’aspiration gastrique douce, intermittente est indispensable afin
de diminuer les effets de l’iléus. Une nutrition parentérale totale est
instaurée. Celle-ci peut être réalisée par cathéter central percutané, cathéter
cutanéo-cave ou Broviac. La suspicion d’une
bactériémie associée nécessite une antibiothérapie
à large spectre. Le recours à des anti-agrégants plaquettaires pour réduire les
micro-thromboses s’est avéré cliniquement inefficace pour certains groupes.
Au stade II, l’état hémodynamique peut
nécessiter l’administration de plasma frais. L’antibiothérapie est systématique
et la surveillance hémodynamique étroite. Les répercussions respiratoires ou
les bradycardies peuvent nécessiter le recours à une ventilation assistée.
Au stade III, l’intervention chirurgicale est
indispensable. Parmi les techniques utilisées citons la double entérostomie de
dérivation, le drainage d’une péritonite et la résection des segments nécrosés.
|
Il va de soi que la surveillance
médico-chirurgicale et radiologique combinée est nécessaire dès le stade I. Sur le plan bactériologique, des cultures de
selles et les hémocultures sont pratiquées. L’antibiothérapie peut consister
soit à l’association d’une pénicilline semi-synthétique ou d’une
céphalosporine à un aminoglycoside ainsi que le recours au métronidazole.
Certaines équipes recourent également à la vancomycine. L’hypoxie et l’hypoxémie doivent être
rigoureusement combattues. Préma 28 semaines, 2 sem de vie.
Ballonnement, rectorragie, bradycardie et hypotension. ASP annexé. Présence
d’un pneumopéritoine ss phrénique gauche et sous hépatique D: ENTEROCOLITE STADE III |
|

Le traitement de l’affection causale doit bien-entendu
être menée de front avec celui de l’entérocolite nécrosante proprement dite.
En dehors du risque de grêle court consécutif à
des résections trop importantes, la complication la plus fréquente à long terme
consiste dans l’apparition de sténoses cicatricielles nécessitant alors des
résections et des entéroplasties ultérieures.
Lors du rétablissement de la continuité grêle ou
colique, l’intégrité du tube digestif est vérifiée par examen en contraste.
Pronostic
Si le pronostic était systématiquement mortel
avant les années 50, il s’est réduit à 50% de mortalité dans les années 80.
Actuellement l’expérience dans les hôpitaux belges approche une mortalité
inférieure à 10%.
L’évolution ultérieure peut également être
conditionnée par les problèmes consécutifs à un poids de naissance très bas.
Rappelons les problèmes nutritionnels, les séquelles de pneumopathie à
membranes hyalines, les hémorragies cérébrales intraventriculaires et le retard
de croissance psychomoteur et staturo-pondéral.
B. Ileus et péritonite méconiale
Le terme d’iléus méconial prête d’emblée à
confusion. Il ne s’agit pas d’un iléus paralytique mais bien d’une obstruction
intestinale mécanique consécutive à l’impaction d’un méconium trop épais. La
composition anormale de ce méconium est pratiquement toujours consécutive à
l’existence d’une mucoviscidose ou fibrose kystique du pancréas. Ce désordre
génétique transmis de manière autosomique récessive peut être dépisté en
période anténatale. L’incidence de la maladie dans une famille présentant des
antécédents est de 25%. Parmi les enfants atteints, 1/5ème d’entre eux
présenteront une obstruction intestinale nécessitant un traitement intensif.
L’affection concernant notamment les sécrétions muqueuses intestinales,
l’épaississement du méconium débute pendant la vie intra-utérine et peut
conduire à plusieurs présentations cliniques :
a) le volvulus de l’anse où a lieu l’impaction
méconiale;
b) une perforation en péritoine libre menant à une
péritonite méconiale éventuellement calcifiée;
c) une souffrance vasculaire menant à une
pseudo-atrésie iléale;
d) une obstruction néonatale souvent grêle
terminale.
Les grands prématurés peuvent présenter un iléus
méconial dont l'origine reste floue. Dans ces cas, aucune anomalie génétique
n'est observée et aucune mucoviscidose clinique n'apparaît.
Les présentations cliniques différentes de la
pathologie méconiale entraînent plusieurs diagnostics très différents:
TABLEAU
|
PRESENTATION |
|
D.D. |
|
PERITONITE MECONIALE |
ANTE OU NEONATALE |
ATRESIE VOLVULUS OMPHALOCELE |
|
OBSTRUCTION |
NEONATALE |
ATRESIE ILEALE |
|
BOUCHON (méconium plug) |
NEONATALE |
HIRSCHSPRUNG MUCOVICIDOSE |
1. Péritonite
méconiale
La péritonite méconiale ou pseudokyste méconial
est classiquement diagnostiquée en période anténatale au cours du dernier tiers
de la grossesse. L’obstétricien ou l’échographiste met en évidence un éventuel
hydramnios, une augmentation du périmètre abdominal et des calcifications
intrapéritonéales. Une masse peut également être observée.
|
Une anamnèse familiale, une ponction
amniotique et une biopsie villositaire conduisent à la confirmation ou à
l’exclusion d’une mucoviscidose. |
Nouveau-né; dystocie par ballonnement
abdominal monstrueux. ASP à la 12ème heure de vie.Images radio-opaques en
piqueté. Peritonite méconiale |
|
|
L’accouchement est programmé dans un centre
d’accueil pluridisciplinaire des malformations congénitales. La césarienne
est hautement conseillée par les chirurgiens pédiatriques. Le traitement est
postnatal immédiat et consiste en une laparotomie. |
|
|

Selon la présentation pseudo-kystique,
péritonéale généralisée ou un éventuel volvulus, la technique chirurgicale
consiste dans un nettoyage de la cavité abdominale, un milking destiné à vider
les anses du méconium anormal et une éventuelle entérostomie permettant une
réalimentation orale ou des irrigations au sérum physiologique et à la
gastrographine. La recherche d’atrésie est systématique et peut être traitée en
première intention ou secondairement. L’évolution postopératoire est
conditionnée par l’atteinte respiratoire.
2. L’iléus
méconial
Cette entité clinique peut survenir dans un
contexte de méconnaissance d’une mucoviscidose. L’enfant présente un ballonnement
abdominal dès l’alimentation, des vomissements bilieux et un empâtement
abdominal caractéristique. Les anses grêles distales peuvent être aisément
palpées donnant à l’opérateur une sensation caoutchouteuse.
Dans certains cas, la présentation peut être plus
aiguë en cas de volvulus.
Les clichés radiographiques standard et
l’échographie abdominale démontrent une raréfaction des images aériques et dans
de rares cas quelques calcifications. L’obstruction est souvent iléale et peut
être confondue avec une atrésie iléale isolée. Un BM test est pratiqué
systématiquement.
La première manoeuvre consiste à mettre l’enfant
sous perfusion et à réaliser un lavement à la gastrograffine; celui-ci confirme
le diagnostic et peut éventuellement traiter l’enfant si le reflux grêle est
suffisant. Le lavement à la gastrographine a également le mérite de
diagnostiquer le bouchon méconial colique gauche. Cette entité
ressemblant à l’iléus méconial consiste en une subocclusion transitoire du
colon gauche spontanément réversible après la vidange du bouchon méconial. En
cas d’échec médical, le traitement de l’iléus méconial peut être
chirurgical. Il l’est systématiquement en cas d’atrésie.
L’intervention peut consister en un simple
milking ou en une résection grêle avec ou sans entérostomies. L’iléostomie en
canon de fusil ou en Y permet des irrigations aisées par les infirmières. Le
pronostic est essentiellement conditionné par l’atteinte générale.
Concernant le diagnostic de mucoviscidose, il
faut rappeler que le test à la sueur n’est pas fiable chez le nouveau-né, après
1 mois le diagnostic peut être finalement confirmé par un test à la
pilocarpine. La réalisation du BM test est également contributive.
TABLEAU-RESUME ILEUS MECONIAL
|
GESTES
IMMEDIATS |
PERFUSION
TOTALE ASP.GASTR.
INTERMITTENTE A.S.P. |
|
|
EXPLORATIONS
COMPL. |
ECHOGRAPHIE LAVT
GASTROGRAPHINE/ACETYLCYSTEINE BM TEST |
|
|
CHIRURGIE |
EXPLORATION
INTESTINALE MILKING-APPENDICECTOMIE (ILEOSTOMIE) (RESECTION
- ENTEROANASTOMOSE) DRAINAGES
(si perforation) RESECTION |
|
C. Les
atrésies duodénales et iléales
Cette entité caractérisée par une clinique
d'occlusion digestive néonatale compose la liste des pathologies obstructives
du nouveau-né.
Les atrésies intestinales constituent avec la
malrotation, les duplications, la maladie de Hirschsprung, l'iléus méconial,
les six principaux diagnostics différentiels du ballonnement abdominal et des
vomissements néonataux.
L'atrésie est l'interruption segmentaire (unique
ou multiple) de la lumière digestive, non pas par son contenu (méconium) mais
de manière intrinsèque. Si la lumière n'est pas totalement obstruée, on parle
de sténose.
L'atrésie
ou sténose duodénale
L'organogenèse du cadre duodénal concerne la
voie biliaire et le pancréas.
Les anomalies d'implantation canalaire du
Wirsung, du Santorini et du cholédoque sont donc associées aux atrésies
duodénales.
|
Avec une incidence de ± 1/2000
naissances, l'atrésie duodénale est souvent diagnostiquée en période
anténatale par la dilatation gastrique en double bulle classique. |
Nouveau-né 12 heures de vie, hydramnios maternel,
ballonnement, vomissements clairs. Hyperventilation et inconfort. Echo non
contributive. ASP annexé. D:
ATRESIE DUODENALE |
|
Un caryotype foetal doit exclure une trisomie
21. A la naissance, les vomissements sont précoces, bilieux en cas
d'implantation sus-atrétique de la voie biliaire, clairs en cas
d'implantation distale de la papille. Le méconium peut être décoloré et rare. Un
ictère peut être associé. L'abdomen est plat et non douloureux. Dès que
l'enfant déglutit de l'air, sans aspiration gastrique, une double bulle
aérique est visible à l'abdomen sans préparation. |
|

Sur le plan clinique, l'association avec une
atrésie de l’oesophage concerne 10% des cas.
L'atrésie duodénale est masquée s'il n'y a pas
de fistule trachéo-oesophagienne (type I).
Le traitement est bien entendu chirurgical. Il
consiste en un by-pass duodéno-duodénal (duodéno-duodénostomie) ou
duodéno-jéjunal (duodéno-jéjunostomie).
Une exploration de la voie biliaire principale
et des voies pancréatique est souhaitable.
Le pronostic est bon en cas de chirurgie
appropriée
L'atrésie
jéjuno-iléale (1-1/1000 à 2000 naissances)
Les formes anatomiques, les sites possibles et
leur nombre sont éminemment variables.
L'étiologie de ces atrésies ou sténoses reste
obscure.
Dans certains cas, les anomalies associées
évoquent un désordre morphogénétiques d'origine génique..
|
Le plus souvent, l'anomalie est isolée et
l'histologie appuie la thèse d'un accident vasculaire ponctuel responsable
d'une nécrose segmentaire . Le diagnostic est souvent anténatal :
polyhydramnios, dilatation intestinale d'amont. |
Garçon
48 h. de vie; ballonnement, pas d'émission méconiale. Vomissements
bilieux. ASP en annexe. Occlusion iléale complète ; Pas d’air
dans le rectum Atrésie jéjunale |
|
Le cycle foeto-maternel de liquide amniotique
est interrompu. Une perforation d'amont causant une péritonite méconiale peut
survenir. Elle entraîne l'apparition de calcifications intrapéritonéales chez
le foetus. |
|


Après la naissance, le tableau clinique est
celui d'une occlusion dont le niveau dépend du site de l'atrésie.
Les vomissements ou l'aspiration gastrique sont
bilieux. L'élévation de la bilirubine indirecte est illustrée par un ictère
chez 1/3 des enfants atteints.
La radiographie d'abdomen sans préparation (ASP)
est pathognomonique. Le principal diagnostic différentiel se fait avec l'iléus
méconial. En effet un retard d'émission méconial (plus de 24h) est observé sur
l'atrésie est iléale terminale.
Un autre diagnostic doit être exclu : une
maladie de Hirschsprung pancolique.
Le diagnostic clinique est souvent évident. Une
opacification du cadre colique jusqu'au-delà de la valvule de Bauhin peut appuyer
le diagnostic clinique.
Le traitement est d'emblée médical (aspiration
gastrique, nutrition parentérale totale et antibiothérapie. La correction
chirurgicale est programmée pour le lendemain; elle consiste à réséquer et
réanastomoser les segments en territoire sain.
L'iléostomie de dérivation est évitée car elle
occasionne des difficultés majeures dans l'alimentation et l'hydratation de
l'enfant. La mortalité est inférieure à 10% et est désormais liée à la
prématurité, immaturité pulmonaire et aux malformations associées.
Il a été rappelé (p. 16) que le tube digestif
vivait de multiples rotations et accolements indispensables.
La première condition inductrice de malrotation
est la croissance intestinale in utero
en dehors de la cavité abdominale. C'est le cas de la hernie diaphragmatique,
de l’omphalocèle ou du gastroschisis. Au plus tôt l'anomalie se manifeste, au
plus elle interfère avec les rotations et accolements secondaires.
La seconde condition regroupe les vices de
rotation au sein de la cavité péritonéale normale. La sémantique est complexe
et dangereuse.
De l'absence totale de rotation de 180° (qui
suit la 1ère
rotation de 90°) correspondant au mésentère commun, au "caecum
flottant", toutes les formes sont décrites incluant défects mésentériques,
hyperrotation...
La
malrotation intestinale
Diagnostic
des obstructions hautes du nouveau-né
· Formes anatomiques variables
· Défaut dans l'un des mécanismes de rotation
et d'accolement pendant la vie embryonnaire
· Associée de fait à tous les défects de paroi
abdominale, diaphragmatiques, duodénaux...
· 4 possibilités![]()
obstruction neonatale
Prise
en charge néonatale
chirurgie
precoce
![]()
volvulus précoce ou tardif
avec isChèmie
réanimation
risque de syndrome
chirurgie
en extrême urgence du grêle court
![]()
troubles chroniques du transit
chirurgie
programmée
coelioscopique ?
![]()
découverte fortuite MESENTERE COMMUN- asymptomatique
surveillance
appendicectomie
COELIOSCOPIQUE
|
Deux variantes anatomiques sont couramment
observées en clinique et doivent être mentionnées : |
Nouveau-né de 6 jours. Vomissements clairs. Abdomen souple. Echo peu contributive. Transit grêle: cliché en procubitus. Ouverture du cadre duodénal ; Treitz à droite de la colonne. Malrotation intestinale |
|
a) Le mésentère commun : symptomatologie
frustre; diagnostic par abdomen sans préparation, échographie et transit. b) La malrotation (type I de Grob) :
symptomatologie est souvent une occlusion duodénale; le diagnostic se fait
par abdomen sans préparation, échographie, transit digestif ou lavement
baryté. Le volvulus grêle peut survenir et sa gravité
implique un traitement préventif. Le tableau clinique est illustré par 4
présentations différentes pour lesquelles chaque stratégie est définie. |
|

La duplication est le dédoublement d'une partie
du tube digestif. La dilatation progressive du segment aveugle conduit à une
compression du segment principal et peut conduire à l'occlusion. Présente à la
naissance, elle peut ne se révéler cliniquement qu'après quelques mois voire
quelques années. La symptomatologie dépend du caractère tubulaire ou kystique.
Le diagnostic est souvent échographique. La
résection du segment atteint est nécessaire. La duplication duodénale forme
avec le cholédococèle (dilatation sacculaire du cholédoque dans la paroi
duodénale) un diagnostic différentiel difficile qui conditionne la technique
chirurgicale.
On parle de dérivation chirurgicale ou de marsupiliation
endoscopique.
|
Enfant
de 18 mois. Subocclusions récidivantes, masse abdominale hyperéchogène à
l'écho. Pas d'altération générale. Transit grêle montrant une compression
extrinsèque. Vue opératoire. D:
Duplication iléale |
|
|

V. LA PATHOLOGIE BILIO-PANCREATIQUE
L'enfant peut être concerné par la pathologie
bilio-pancréatique. Les affections hépato-biliaires pédiatriques sont
sensiblement différentes de celles de l'adulte (cirrhose éthylique, lithiase
vésiculaire, pancréatite aiguë, ...).
On distingue :
A. L'atrésie des voies biliaires
B. La dilatation congénitale des voies biliaires
C. La lithiase vésiculaire
D. Les tumeurs et abcès hépatiques
E. L'hypertension portale
F. La mucoviscidose
G. La pancréatite idiopathique
A. L'atrésie des voies biliaires
Rare (1/10.000 naissances), l'atrésie des voies
biliaires apparaît de plus en plus comme une affection acquise in utero
(infection virale foetale par rétro virus 3) conduisant à une fibrose
canaliculaire intra- ou extra-hépatique.
Le fait clinique essentiel est l'apparition ou
le maintien d'un ictère néonatal intense avec hyperbilirubinémie conjuguée et
hépatomégalie.
Le diagnostic différentiel de l'ictère néonatal
interpelle le pédiatre et chirurgien.
Le tableau en annexe mérite une analyse.
Il reprend les examens qui permettent de
progresser dans l'évaluation d'un ictère néonatal (Coombs, bilirubinémie,
échographie des voies biliaire, laparotomie-cholangiograhie).
L'évolution naturelle conduit à l'hypertension
portale, la malnutrition et le décès autour d'un an.
Lorsque l'atrésie des voies biliaires concerne
essentiellement les voies biliaires extra-hépatiques, la malformation est
considérée comme chirurgicale corrigible. Une hépatico-entérostomie par anse
montée et anastomose bilio-digestive peut en effet être pratiquée; celle-ci
retarde de 10 à 20 ans la survenue de l'insuffisance hépatique terminale.
Secondairement, une transplantation hépatique peut être envisagée.
Lorsque l'atrésie atteint les canalicules biliaires,
l'hépatico-entérostomie n'est pas fonctionnelle. La seule chance consiste alors
à greffer un lobe hépatique (donneur décédé ou vivant) à l'enfant atteint. La
transplantation doit être effectuée autour de l'âge d'un
an. L'affection est grave, le pronostic réservé et le traitement coûteux et
restreint à quelques centres de pointe occidentaux.
B. Dilatation congénitale des voies biliaires
Communément appelée kyste congénitale du
cholédoque, la dilatation de la voie biliaire regroupe en fait plusieurs
formes. La classification d'Alonso Lej de 1959 est encore la plus connue.
Quatre types sont rencontrés classiquement chez l'enfant comme chez l'adulte :
· La dilatation de la voie biliaire principale
(type I).
· Le kyste pendulaire implanté latéralement sous
le confluent cholédoco-cystique (type II).
· Le cholédococèle (type III).
· La dilatation intrahépatique (type V) ou
maladie de Caroli.
L'étiologie reste obscure et controversée
(hypothèse de Babbit).
La symptomatologie est variable :
·
Ictère
·
Lithiase biliaire
·
Pancréatite
·
Masse palpable
·
Angiocholite
Le diagnostic est réalisé par échographie,
cholangio- rétrograde endoscopique (ERCP), CT scanner, scintigraphie hépatique.
Le traitement est impératif et chirurgical. Il
consiste à réséquer la dilatation (risque de cancérisation) et pratiquer une
anastomose bilio-digestive. Seule la maladie de Caroli nécessite une prise en
charge médico-chirurgicale associant drainage endoscopique (angiocholite) et
résection chirurgicale (cirrhose biliaire secondaire).
C. La lithiase vésiculaire
Elle concerne le grand enfant, plutôt la fille
et majoritairement les enfants atteints de maladies hémolytiques
(drépanocytose...).
Le diagnostic s'appuie sur la clinique (douleurs
postprandiales, cholécystite aiguë, pancréatite biliaire, angiocholite..) et
l'échographie.
Le prématuré alimenté par voie parentérale peut
présenter un sludge vésiculaire avec cholécystite secondaire.
Le traitement est chirurgical et consiste en une
cholécystectomie par voie coelioscopique.
D. Tumeurs et abcès hépatiques
La tumeur hépatique la plus fréquente est
l'hépatoblastome. L'étudiant se référera au cours de Gastro-entérologie et
Oncologie.
Parmi les masses néoplasiques hépatiques, les
métastases d'une tumeur de Wilms ou d'un neuroblastome sont en général
découvertes dans le décours du follow-up d'une autre intervention chirurgicale.
Les abcès parasitaires existent chez les
enfants. Leur pathogénie et leur traitement sont semblables à ceux de l'adulte
(voir cours de Chirurgie digestive de 1er Doctorat). L'amibiase touche les
enfants africains. A côté du traitement par métronidazole, le chirurgien peut
être amené à drainer certains abcès.
Le kyste hydatique est dû
à l'echinoccoque . Le diagnostic est échographique ou tomodensitométrique et confirmé
par la recherche d'anticorps anti-échinoccoque.
Par laparotomie (peut-être par coelioscopie) le
traitement consiste à vider le kyste au milieu de solution NaCl 3 %, et à
réséquer la membrane proligère. La périkystectomie est défendue parfois. Les fistules
biliaires doivent être suturées. Cette pathologie est moins souvent rencontrée
à Bruxelles qu'il y a 15 ans.
E. L'hypertension portale
L' hypertension portale (HTP) consiste en une
altération du flux portal suite à un obstacle pré-, hépatique ou post-. La
plupart des données physiopathologiques de l'adulte sont transposables à
l'enfant. Parmi les différences, on relève que le flux sanguin hépatique dépend
proportionnellement plus du débit portal que du débit artériel hépatique.
L'étiopathogénie est spécifique chez l'enfant.
La première cause chez le nourrisson est la
thrombose préhépatique sur phlébite ombilicale au départ d'un sepsis sur
cathétérisme. Après une phase d'hypertension mésentérique, les collatérales du
pédicule hépatique se développent et forme un panier veineux erronément appelé cavernome de la veine porte.
La deuxième cause est la cirrhose sur atrésie
des voies biliaires intra-hépatiques; elle peut également suivre l'intervention
de Kasaï après atrésie extra-hépatique.
La fibrose hépatique, le déficit en alpha 1
antitrypsine et les syndromes d'hypercoagulation sont plus rares.
Présentation
clinique
La symptomatologie survient en principe
graduellement dans les pays occidentaux. Elle est souvent l'aboutissement d'une
insuffisance hépatique.
On découvre une hématémèse, un méléna, une
anémie chronique ou une hépatosplénomégalie. L'ascite, la circulation veineuse
sous-cutanée abdominale (tête de méduse) sont pathognomoniques.
Explorations
et bilan
A part les examens hématologiques complets (Hb,
Hcte, V.G, V.S., PTT,...) on réalise une échographie, un bilan fonctionnel
hépatique.
Après l'âge de 6 mois, l'endoscopie haute
peut-être pratiquée sous narcose.
La splénoportographie percutanée permet de
réaliser une cartographie veineuse et d'envisager les possibilités de
dérivation chirurgicale.
Prise
en charge d'une hématémèse de l'enfant
La complication la plus classique et la plus
grave de l'HTP est l'hématémèse. Le tableau est spectaculaire et dramatique
pour l'enfant et ses parents. La pâleur et la tachycardie sont brutales, le
vomissement sanglant est puissant et répétitif.
L'enfant doit être immédiatement transporté en
S.A.M.U. vers un centre équipé en endoscopie opératoire pédiatrique.
Le traitement immédiat est le suivant:
·
Transfusion sanguine
·
Sédation légère
·
Position semi-assise
·
Monitoring complet
·
Endoscopie sous narcose et sclérothérapie
immédiate
·
Vitamine K et Calcium
·
Somatostatine
·
Antibiothérapie prophylactique (
Céphalosporines)
On évitera la sonde gastrique, la
sonde de Blakemore, et la broncho-pneumonie de déglutition en intubant l'enfant
en cas de choc hypovolémique.
Le traitement
de fond est le suivant:
·
Sclérothérapie préventive
·
Shunt spléno-rénal
·
Shunt mésentérico-cave
·
TIPS chez le grand enfant
·
Transplantation hépatique si insuffisance
hépato-cellulaire
E.
LA
MUCOVICIDOSE
La
mucoviscidose appelée en anglais « cystic
fibrosis » est une affection génétique aux implications métaboliques
et biochimiques nombreuses. Cette affection congénitale décrite largement dans
les cours de génétique, biochimie et anatomie pathologique est développée
cliniquement dans le cours de pédiatrie. Comme ses complications digestives et
respiratoires concernent le chirurgien pédiatrique, la maladie a été évoquée
dans le diagnostic différentiel des occlusions digestives du nouveau-né.
Chez
le plus grand enfant, le gastro-entérologue peut être confronté à/
·
une
insuffisance pancréatique exocrine conduisant à une
malabsorption des graisses et une carence lipidique compliquée d’un retard de
croissance. Le traitement substitutif consiste à administrer des formes
gastro-protégées de pancréase et créon. Un supplément vitaminique
liposoluble est utile (A D E K ). Il est également recommandé de supplémenter
en Fe, Cu, Zn et Se.
·
une
constipation chronique avec impaction fécale, prolapsus et
parfois une occlusion digestive basse aiguë.
·
une
cardiomyopathie et un cœur pulmonaire chronique
concernent le chirurgien cardiaque pédiatrique spécialisé en transplantation
cardio-pulmonaire. En effet, il faut préférer la transplantation pulmonaire en
première intention.
F. LA
PANCREATITE CHRONIQUE IDIOPATHIQUE
La littérature foisonne d’hypothèses au sujet de
la pancréatique chronique et de sa forme infantile. Les faits se limitent à une
inflammation canalaire associée à une modification de la chimie des sécrétions
pancréatiques entraînant des protein
plugs et des sténoses canaliculaires puis canalaires. Des calcifications
intraparenchymateuses apparaissent. L'insuffisance exocrine puis endocrine
suit.
Les douleurs abdominales postprandiales
récurrentes et intenses sont le signe d'appel. La perte de poids, la
stéatorrhée et le diabète sont plus tardifs.
La mise au point est biologique, échographique.
La pancréatographie endoscopique est indiquée si une dilatation ou endoprothèse
s'impose.
En cas de dilatation du canal de Santorini ou de
Wirsung, une pancrético-jéjunostomie est utile. Le principe est de conserver le
plus de parenchyme pancréatique possible. Il s'agit d'un traitement
médico-chirurgical réservé à des centres spécialisés.
VI. LES TROUBLES DE LA MOTRICITE DIGESTIVE
Les quatre pathologies qui composent ce chapitre
étaient jadis totalement séparées dans les chapitres cliniques auxquels leur
symptomatologie les réfèrent. Le tableau suivant les cite:
TABLEAU 10
|
Pathologie |
Age |
Symptôme
dominant |
Chapitre
clinique |
|
REFLUX GASTRO-OESOPHAGIEN |
0-Adulte |
Asthme Pyrosis |
Maladies respiratoires Pathologie oeso-gastrique |
|
ACHALASIE
OU MEGAOESOPHAGE |
15-Adulte |
Dysphagie |
Pathologie oesophagienne |
|
STENOSE
HYPERTROPHIQUE DU PYLORE |
2-6 semaines |
Vomissements |
Occlusions hautes post-natales |
|
MALADIE
DE HIRSCHSPRUNG et
dysganglionoses |
0-Adulte |
Constipation |
Occlusions basses, constipation |
En fait, ces pathologies sont consécutives à un
trouble intrinsèque de la motricité digestive. Le relais physiopathologique et
histochimique de chaque entité clinique fait appel à un désordre histologique
et histochimique au niveau de l'innervation sympathique et parasympathique du
tube digestif . A côté des anomalies structurales morphologiques, on découvre
de profondes anomalies au niveau des neurotransmetteurs . Le lecteur est invité
à se référer aux cours d'anatomie pathologique et d'histopathologie. Le
chirurgien pédiatrique se doit de suivre les recherches dans ces domaines pour
affiner le diagnostic et améliorer la thérapeutique ou sa séquence;
A. L'achalasie du cardia ou megaoesophage
Il
faut plusieurs années pour que l'oesophage se dilate en amont d'un cardiospasme. Pendant la déglutition,
des complexes moteurs coordonnés se succèdent le long des 2/3 distaux de
l'oesophage. Le cardia doit s'ouvrir en phase pour permettre au bol alimentaire
de pénétrer dans l'estomac. Si les médiateurs relaxant le muscle du cardia sont
inefficaces ou que la neurotransmission est déficiente au niveau de la jonction
neuro-musculaire, le cardia reste fermé. La musculature oesophagienne
s'hypertrophie et la stase oesophagienne apparaît.
La symptomatologie est progressive mais conduit
à des érosions muqueuses.
Le diagnostic est confirmé par un transit
oeso-gastrique baryté, une endoscopie ou surtout une manométrie qui distingue
plusieurs formes. Il existe la forme classique sans relâchement cardial isolé,
et des formes plus complexes comme la présence d'ondes oesophagiennes
spastiques non coordonnées conduisant au nutcracker
syndrome.
Dans l'achalasie du cardia de l'adolescent ou de
l'adulte jeune on peut être obligé de réaliser une myotomie selon Heller. La
dilatation pneumatique endoscopique suffit souvent à soulager le patient pour
une longue durée.
B. LE REFLUX GASTRO-OESOPHAGIEN
Le reflux de liquide gastrique vers l'oesophage
survient pratiquement chez tout individu à un moment de sa vie. Certains
prétendent même qu'il fait partie de la physiologie normale du nouveau-né ou de
la femme enceinte même s'il est asymptomatique. Dans un cours de pathologie,
nous nous limiterons au reflux invalidant, symptomatique inducteur de
complications respiratoires, oesophagienne ou accompagné d'anomalie anatomique
comme une hernie hiatale.
A la naissance, la jonction cardio-tubérositaire
ne correspond pas à l'anatomie du cadavre enseignée en candidatures en sciences
médicales. Tout d'abord, il y a immaturité des plexus myentériques et ouverture
de l'angle cardio-tubérositaire; le diaphragme est prédominant pendant
l'inspiration et il ne subit pas de la musculature abdominale une
contre-pression puisque cette musculature est hypotrophique.
De plus, après une tétée ou un biberon,
l'estomac contient une quantité impressionnante d'air qu'il doit régurgiter en
ouvrant son cardia.
Symptomatologie
Là où le médecin intervient c'est lorsque le
phénomène est exagéré, entraînant de véritables vomissements postprandiaux et
des régurgitations incessantes accompagnées d'inconfort, d'irritabilité, de
troubles respiratoires tels que l'asthme et la toux nocturne.
Les incidents respiratoires peuvent être masqués
par une composante allergique qui est en fait compliquée d'un R.G.O.
On peut voir des pneumonies à répétition chez
les petits enfants et une oesophagite exulcérée chez le plus grand enfant (en général)
Explorations
et bilan
L’anamnèse et l'observation de l'enfant
éclairent le praticien mais le diagnostic reste délicat.
Actuellement la phmétrie de 24 heures permet de
mesurer le nombre d'épisodes où le pH oesophagien chute sous 4 et sa corrélation
clinique. Le pourcentage de temps discrimine le R.G.O. pathologique du
physiologique.
A cette phmétrie, peut être associée une
manométrie oesophagienne
La radiologie montre s'il
existe une hernie hiatale ou une béance.
La présence d'une vraie hernie hiatale ne
présage pas d'une bonne réponse médicale et suscite l'indication d'une
correction chirurgicale définitive.
En cas de sténose, d'hématémèse, une
oesophagoscopie peut être faite.
Traitement
Méthodes
conservatrices:
précautions
posturales
diététique
kinésithérapie
respiratoire
Médicaments:
prokinétiques
(prépulsid, métoclopromamide)
anti
H2 (cimétidine,
ranitidine....)
I.P.P.
(oméprazole, lansoprazole...)
Chirurgie: Nissen ou Toupet
coelio
ou laparotomie
C. LA STENOSE HYPERTROPHIQUE DU PYLORE
La Sténose Hypertrophique du pylore (SHP)
constitue la plus fréquente indication chirurgicale en pratique quotidienne
chez le nourrisson.
Décrite depuis 1646 par Hildanus, et étudiée par Hirschsprung(!) en 1888, l’hypertrophie
néonatale évolutive du muscle pylorique fut considérée comme responsable de
l’occlusion gastrique survenant de 2 à 6 SEMAINES. La couche circulaire
hypertrophiée comprime la muqueuse qui finit par se collaber totalement.
|
|
coupe sagittale dans une olive pylorique la muqueuse est collabée par l’hypertrophie de
la musculeuse cet aspect est parfaitement rendu par l’image
échographique |
|
|
|

L’estomac se dilate, s’hypertrophie et reflue
(R.G.O.).
Les anomalies intrinsèques ont été étudiées.
Quelles sont les certitudes?
L’anomalie ne touche que les nourrissons ( 350/
an en Belgique), souvent les garçons (sex ratio 3/1), plutôt les Européens
blancs, jamais les Chinois.
L’incidence familiale soutend le rôle génétique,
l’incidence saisonnière (automne et hiver), sociale et le rôle du stress
maternel illustrent le contexte environnemental.
La présence d’une hypergastrinémie et son rôle
n’est pas clairement démontré.
Symptomatologie
Les vomissements répétés en jets, la
déshydratation et la palpation d’une olive pylorique constituent la triade
classique.
Les parents notent une stagnation pondérale
voire une perte de poids en phase avancée; l’enfant se constipe, un ictère
apparaît.
La gluconyl-transférase hépatique est effondrée,
la chute de l’apport calorique et une compression mécanique des voies biliaires
extra-hépatiques sont invoquées.
Le médecin trouve un enfant assoiffé et avide de
téter, alors qu’il ne tolère pas son biberon. Après le repas, l’estomac se
contracte; les ondes sont perceptibles chez l’enfant déshydraté. Pendant la
tétée, l’olive peut être palpée devant la colonne avec la main gauche.
Explorations
et bilan
L’anamnèse et l'observation de l'enfant
éclairent le praticien.
La biologie traque l’alcalose et l’hypokaliémie.
L’A.S.P. montre la dilatation gastrique caractéristique. L’échographie confirme
l’indication opératoire.
Le transit baryté est proscrit. D’abord il
risque de favoriser une pneumopathie de déglutition et ensuite il gêne le
chirurgien.
La sténose hypertrophique du pylore est
l’occasion de présenter un algorithme des diagnostics devant un vomissements
chez le nourrisson.
Traitement
L’enfant est perfusé avec su sérum I/2
physiologique et des ions K+; une sonde gastrique peut être placée en cas
d’inconfort. Le jeun strict est imposé.
Les Scandinaves ont tenté et réussis à traiter
certains enfants sans les opérés. Le principe est d’instituer une nutrition
parentérale ou par feeding transpylorique et d’attendre. Après 3 à 5 semaines,
l’hypertrophie régresse, le défilé pylorique s’ouvre et l’enfant guérit. Comme
on peut s’y attendre, ni les parents ni l’INAMI, ni les chirurgiens
n’approuvent ce traitement.
Après réhydratation, le chirurgien peut opérer.
On peut donc affirmer que le traitement de
référence est la pylorotomie extra-muqueuse par voie mini-invasive ombilicale.
Le muscle pylorique est incisé longitudinalement
sans effracter la muqueuse. Douze heures plus tard, on réalimente l’enfant qui
sort généralement au 3ème jour post-opératoire.
Un R.G.O. peut persister quelques semaines
D. LA MALADIE DE HIRSCHSPRUNG
La maladie de HIRSCHSPRUNG ( Mégacôlon
congénital ) est une aganglionose colique. Elle constitue la forme la plus
caractéristique des anomalies de l’innervation digestive qui s’exprime chez
l’enfant.
L’origine fondamentale du déficit reste obscure
mais plusieurs observations histologiques, biochimiques, neurophysiologiques et
embryologiques éclairent le clinicien.
Histopathologie:
Les cellules ganglionnaires des plexus
myentériques ss-muqueux (Meissner) et intermusculaires (Auerbach) sont quasi ou
totalement absentes. Les fibres nerveuses
pré-synaptiques non myélinisées se perdent de manière anarchique au sein
des fibres intrinsèques sans jonction synaptique fonctionnelle.
L’acétylcholine est le principal médiateur, ce
qui conduit à dépister l 'anomalie par un test à la cholinestérase. Si les
fibres adrénergiques sont pratiquement absentes, on note une concentration
anormale de substance P, PYY et VIP.
L’anatomie de la zone transitionnelle est encore
plus complexe. Pour le clinicien, il résulte surtout que la reconstruction
rectale et les dérivations éventuelles
ne doivent utiliser que le colon normal, voire le grêle en cas de Hirschsprung
pancolique.
Neurophysiologie
L’atteinte du sphincter anal interne (S.A.I.)
par la maladie contrarie évidemment la physiologie de la défécation et
l’appareil de continence et d’exonération.
Par exemple, le réflexe recto-anal inhibiteur
est absent. La dilatation d’un ballonnet intra-rectal ne déclenche pas la
contraction du S.A.I.
Génétique
L’incidence est de 1 pour 5OOO naissances soit
prés de 30 cas en Belgique par an. Les garçons sont plus souvent atteints.
L’atteinte de jumeaux, l’association (2%)
rare mais significative avec des anomalies chromosomiques argue pour une
étiologie génétique qui freinerait la migration des cellules de crêtes neurales
par trouble moteur cellulaire
intrinsèque ou par réaction immunitaire cellulaire.
Symptomatologie
Le retard d’émission du méconium et une
constipation néonatale sont les principaux stigmates. Le nouveau-né présente un ballonnement rapide, une surélévation
diaphragmatique, des vomissements et une dénutrition rapide. L’A.S.P. montre
une dilatation colique en amont d’un segment recto-sigmoïdien plus ou moins
long collabé contenant des selles. L’occlusion se répercute sur le grêle. Si la
forme est pancolique, elle peut être confondue avec un iléus méconial ou une
atrésie.
Cependant, plusieurs formes existent:
La présentation subaiguë:
L’enfant sort de la maternité et présente une constipation
d’installation progressive.
Un retard de développement staturo-pondéral et
même un marasme (dans les régions économiquement peu développées - l’Afrique
centrale ou appauvries - région du Centre) accompagnent les plaintes
abdominales.
La présentation septique:
L’entérocolite et le transfert de la flore
bactérienne conduit à la septicémie. Masqué par une éventuelle autre pathologie
(hernie, prématurité, ....) le tableau se complique d’un sepsis souvent grave
et mal élucidé. Le choc et l’insuffisance respiratoire aiguë conduisent
d’ailleurs à une mortalité significative. Le tableau abdominal peut alors être
faussement interprété comme secondaire au sepsis et non primitif.
La présentation tardive:
Il s’agit plutôt de dysganglionoses mais le
diagnostic peut passer inaperçu pendant plusieurs années. Il est même parfois
posé chez l’adulte après des mois ou des années de lavements répétés.
HIRSHSPRUNG
NEONATAL: diagnostic différentiel:
· ILEUS
ET BOUCHON MECONIAL
·
VOLVULUS
·
ATRESIE
ILEALE
·
ENTEROCOLITE
NECROSANTE
·
SEPSIS
·
HYPOTHYROIDIE
·
HEMORRAGIE
SURRENALIENNE
· HYPOKALIEMIE
·
Explorations
et bilan
Toucher rectal
Le toucher rectal réalisé avec prudence permet
d’examiner le canal anal, de provoquer éventuellement une expulsion fécale.
ASP
On peut rechercher une déshabitation, un
encombrement stercoral mais surtout une dilatation colique d’amont.
Lavement sans préparation
La disparité de calibre colique signe le
diagnostic; il faut se méfier des microcolons congénitaux.
Biopsie rectale
Par aspiration, chirurgicalement sous narcose ou
par sphinctéromyotomie endo-rectale ( parfois même sur appendicectomie!),
l’examen permet une analyse extemporanée, une coloration argentique et une
étude histochimique.
Manométrie ano-rectale
Elle complète la mise au point mais n’est pas
indispensable.
Traitement
Si la présentation est frustre, on commence souvent par des lavements évacuateurs. Si
l’enfant présente une occlusion aiguë, la mise en perfusion et l’aspiration
gastrique sont nécessaires. Le chirurgien réalise une biopsie rectale puis une
dérivation intestinale en territoire sain. L’iléostomie est défendue par
certains. Après la phase aiguë, souvent après 6 mois, on réalise une
proctocolectomie avec reconstruction rectale selon SOAVE, DUHAMEL, LESTER
MARTIN (H. Total) ou SWENSON. De nouvelles variantes sont décrites dont la
mucosectomie endorectale et l’anastomose colo-anale. Certains réalisent la
colectomie par coelio-chirurgie.
L’enfant sera suivi de près jusqu’à sa
continence. Les sténoses ou incontinences sont parfois objectivées.
VI. LES ANOMALIES DE LA PAROI ABDOMINALE
Comme cela fut expliqué et enseigné dans le
cours de chirurgie digestive, la paroi
abdominale est le siège d’anomalies, de
traumatismes et d’affections en général auxquels le médecin généraliste, le
chirurgien ou le pédiatre sont très souvent confrontés.
Du diaphragme au carrefour inguinal, les
anomalies morphogénétiques touchent plus volontiers certains sites critiques ou
de multiples diagnostics différentiels se rencontrent. Certains auteurs
classifient ces pathologies selon le site (l’ombilic, le creux inguinal....),
d’autres selon les plaintes (vomissements, douleurs abdominales
chroniques,...). Nous vous proposons d’exclure du chapitre la pathologie
diaphragmatique que nous avions choisie pour illustrer la prise en charge des
malformations congénitales graves et de regrouper les autres pathologies dans
ce chapitre. La présentation clinique prévaudra:
1.
Les défects ombilicaux courants et bénins
s’ils sont reconnus et traités
2.
Les grands défects antérieurs
diagnostiqués in utero et de pronostic vital
3.
Le carrefour inguinal siège de multiples
et passionnants diagnostics différentiels.
Embryologie
Pour comprendre l’étiologie, l’anatomie et la
clinique de ces anomalies, il est fait appel aux aspects morphogénétiques que
le futur médecin doit se rappeler.
L’ébauche d’une paroi abdominale antérieure
n’est pas évidente en examinant le disque trilaminaire que constitue un embryon
de 3 semaines. En fait la cavité coelomique apparaît par le repli céphalocaudal
et latéral du mésoderme et de l’ectoderme autour d’un centre fictif : l’anneau
ombilical. A travers ce défect programmé et incontournable, passent les
vaisseaux ombilicaux, le résidu vitellin (canal omphalo-mésentérique) et le
résidu allantoïdien (ouraque) , le tout recouvert de la membrane amniotique. La
croissance et l’épaississement du feuillet mésodermique constitue une enveloppe
solide qui ne respecte qu’un petit passage autour du cordon ombilical. Tous les
problèmes cliniques antérieurs sont liés à ce processus.
Par anomalie génique (gène codant pour une
quelconque propriété de migration ou de fusion cellulaire) ou accident
tératogène (hémorragie traumatique, choc - hypoxémie tissulaire...) les
malformations sont diverses et encore obscures.
En ce qui concerne la fermeture de la sangle
pelvienne et du canal inguinal, le lecteur se rappellera la descente des
gonades et la sortie anténatale du testicule par l’orifice profond et externe
qui laisser passer les vaisseaux spermatiques et le canal déférent. La
musculature enserre l’orifice et la cicatrice du refoulement péritonéal (canal
péritonéo-vaginal) disparaît par collapsus; la vaginale est dès lors séparée de
la cavité coelomique. La cryptorchidie, l’hydrocèle, la hernie inguinale et le kyste
du cordon sont des formes différentes de l’échec de ce processus
morphogénétique.
A. les
hernies ombilicale et épigastrique ET AUTRES
PATHOLOGIES ombilicales
La hernie ombilicale (HO) est une voussure
centrée autour de l’ombilic et
recouverte de
peau. Elle peut être décelée dès la naissance ou se développer
au départ d’un tout petit défect ignoré à la
naissance.
|
Plus fréquente dans la race noire, la HO
disparaît classiquement avant 18 mois lors de la fusion des grands droits. Si l’enfant présente un diastasis des grands |
Fillette
à 10 jours de vie, protrusion centrée sur l'ombilic. Pas de fièvre, pas de
plainte digestive. D:
Hernie ombilicale |
|
droits ou une HO dont le diamètre dépasse 2
cm,, il y a peut de chance de la voir se refermer spontanément, surtout si
après la marche, le médecin constate qu’elle s’élargit. Le risque d’étranglement et d’obstruction digestive est minime (contrairement aux adultes) . Les parents
européens n’apprécient pas son esthétique et demandent une correction chirurgicale. |
|

L’intervention consiste à réséquer le sac et
fermer la ligne blanche par une mini-incision péri-ombilicale.
Le collage d’un dollar ou de 20 francs qu’osent
encore préconiser certains médecins généralistes s’apparente à la sorcellerie
mais pas à la médecine rationnelle et scientifique.
La hernie
épigastrique de la ligne blanche est médiane et
asymptomatique. Elle est entourée d’un anneau fibreux . Elle apparaît lors du
Valsalva et ne guérit jamais spontanément. La grande fille demande parfois sa
correction chirurgicale pour raisons esthétique. La cure chirurgicale laisse
malheureusement une cicatrice.
Les résidus
omphalo-mésentériques correspondent à la fermeture incomplète
ou à une anomalie de finition et de différenciation des annexes foetales et du midgut . On distingue:
· le
sinus omphalo-mésentérique
· la
fistule omphalo-mésentérique
· le
diverticule de Meckel
· le
sinus de l’ouraque
· le
granulome ombilical
L’examen physique et l’inspection détaillée sont
indispensables: le clinicien recherche un écoulement de liquide séreux,
purulent ou urinaire; l’ombilic est examiné à la recherche de muqueuse; un
trajet fistuleux est éliminé. L’échographie permet de dépister ou confirmer un
kyste, une collection ou un trajet organisé, connecté à la vessie ou au grêle.
Une fistulographie, une cystographie et un transit grêle peuvent être demandés
dans les formes complexes requérant une laparotomie ou une coelioscopie.
L’évolution naturelle et les complications sont
variables.
L’infection du sinus omphalo-mésentérique
nécessite un drainage et éventuellement une antibiothérapie. Le danger d’une
omphalite du nouveau-né doit être connu et écarté car elle peut entraîner une
thrombose portale. Le granulome peut être électrocoagulé ou nitraté. Les
résidus muqueux ectopiques doivent être chirurgicalement réséqués. En cas de
fistule (avec un Meckel par exemple) le chirurgien doit réaliser une
intervention complète.
Il en est de même pour les kystes de l’ouraque
(voir cours d’Urologie).
B. L’OMPHALOCELE
ET LE GASTROSCHISIS
Les malformations majeures de la paroi
abdominales sont diagnostiquées normalement par l’obstétricien ou
l’échographiste anténatal. La gravité du défect, le sepsis, l’hypothermie et
les malformations associées entraînent le décès du nouveau-né en l’absence de
traitement. Malgré leur apparente ressemblance, ces deux malformations
congénitales présentent d’importantes différences.
1
LE GASTROSCHISIS
Le foetus présente une éviscération partielle
par un orifice latéro-ombilical souvent droit. Les anses intestinales baignent
dans le liquide amniotique et sont recouvertes de dépôts amniotiques. Il s’agit
en général d’anses grêles. L’intestin n’est bien évidemment pas fixé aux
gouttières pariétocoliques et la malrotation peut entraîner un volvulus
anténatal ou néonatal. L’ischémie mésentérique localisée entraîne des atrésies
ou des sténoses.
Il n’y a pas d’anomalie génétique démontrée.
A la naissance,
une déperdition liquidienne et calorique mènent
à la déshydratation, à l’acidose et à la péritonite.
TABLEAU 11
Distinction
entre le gastroschisis et l’omphalocèle
|
Observation |
GASTROSCHISIS |
OMPHALOCELE |
|
Localisation |
Latérale au cordon |
Centrée , surplombée par le cordon |
|
Taille |
2 - 4 cm |
2 -10 cm |
|
Couverture |
Aucune |
Sac amniotique |
|
Contenu |
Grêle (colon) |
Intestins, Foie, pancréas, gonades... |
|
Intestin |
Epaissi, couenneux |
normal |
|
Malrotation |
OUI |
OUI |
|
Motricité |
Iléus prolongé |
Normale |
|
Anomalies
digestives |
Atrésies |
Rares |
|
Syndromes |
Beckwith, Trisomies 13/15... |
Inconnu |
|
Réduction
chirurgicale |
Aisée |
Difficile ou impossible |
|
Fermeture
défect |
Aisée |
Difficile ou retardée |
2.
L’OMPHALOCELE
L’omphalocèle est la protrusion de viscères
intra-abdominaux en dehors de la cavité abdominale par un défect centré sur
l’ombilic et surplombé par l’implantation des vaisseaux ombilicaux.
Les
viscères sont entourés d’une membrane avasculaire épaisse constituée
de l’amnios et de la gelée de Wharton; après 3 jours, la membrane s’opacifie,
se nécrose et se délabre, laissant les viscères à l’air. L’infection survient
alors rapidement et conduit au sepsis et au décès.
Prise
en charge
Comme cela fut expliqué, le diagnostic doit être
anténatal . La mère et le père sont pris en charge par un centre
multidisciplinaire qui réalise une ponction au cordon pour caryotype foetal,
une anamnèse familiale et un premier conseil anténatal. Celui-ci se
transformera en un conseil génétique complet et formel après la naissance ou
l’interruption de grossesse éventuelle.
L’accouchement est programmé et déclenché dans
un hôpital universitaire où l’équipe néonatale médico-chirurgicale prend le
nouveau-né en charge. La fin de la grossesse est suivie afin d’obtenir la
meilleure maturation pulmonaire mais en évitant de laisser évoluer par exemple
un volvulus. Il n’y a actuellement aucune indication de chirurgie anténatale.
A la naissance, l’enfant est aspiré au niveau
trachéal et gastrique. Il est perfusé et emballé immédiatement dans un sac
étanche et imperméable. Le chirurgien pédiatrique doit être présent auprès de
l’anesthésiste et du pédiatre.
Une attention particulière est portée aux
besoins hydriques de 175 ml/Kg. j. 10 % dextrose en lactate Ringer. Un
cathétérisme vésical est nécessaire et une couverture antibiotique est
prophylactique (Ampi/ Amikacine).
Plusieurs techniques chirurgicales coexistent.
Le gastroschisis est souvent exploré et réintégré en première intention. Un
stretching permet aux muscles d’être suturé en un plan. La peau recouvre le
défect. Pendant la réintégration, l’anesthésiste monitorise la pression
intra-gastrique et les paramètres vitaux afin d’éviter une augmentation de la
pression intra-abdominale au-delà de 15 cm H2O. Le retour cave et la
distension diaphragmatique risque de diminuer le débit cardiaque, d’accroître
les pressions d’insufflation voire d’induire une hypoperfusion rénale ou une
thrombose veineuse.
Il ne servirait à rien de réintégrer un grêle
accompagné d’une thrombose veineuse mésentérique.
Dans les grands défects, souvent l’omphalocèle,
il est nécessaire de recourir à la technique du SILO ou Schuster technique. Une
enveloppe de polypropylène transparent est suturée autour du défect. Le
réservoir constitué est suspendu à l’apex et une traction est opérée pendant
quelques jours. Dans un deuxième temps, après ventilation et curarisation, il
devient possible de réaliser un nouveau stretching. La poche est enlevée et une
suture musculo-cutanée est réalisée.
L’enfant est traité par nutrition parentérale
totale pendant plusieurs jours ou quelques semaines tant que l’iléus ne s’est
pas amendé. De multiples stratagèmes sont enseignés en post-graduat aux futurs
chirurgiens.
En dehors des complications liées aux anomalies
digestives éventuelles, l’opéré peut présenter une entérocolite lors de la
réalimentation orale. La qualité du nursing influe aussi sur le
pronostic vital de ses enfants.
C. LE
CARREFOUR INGUINAL.
Le terme carrefour est justifié par la réunion
sur un même site anatomique d’une multitude de pathologies de paroi, digestives
et urinaires. Aux confins de plusieurs disciplines connexes et en parallèle
avec l’enseignement d’urologie et de gastro-entérologie, nous allons tenter
d’éclaircir le diagnostic différentiel de cette région.
L’anatomie est connue. La morphogenèse comprise.
LA
HERNIE INGUINALE
Il s’agit d’un des motifs les plus fréquents de
consultation.
La pathologie affecte 2 % des garçons et 0.3 %
des filles. La pathologie est bénigne, facilement identifiable, jamais
spontanément résoluble et cliniquement décelée dès que le sac herniaire se
remplit. Bénigne à froid, elle se complique de manière imprévisible d’étranglement
et d’occlusion digestive. Non opérée elle peut conduire à la nécrose ovarienne
chez la fille ou à la péritonite. Elle peut être associée à une cryptorchidie .
La prématurité expose le nouveau-né à un risque atteignant 30 %.
La pathogénie est assez bien identifiée. Au
cours du troisième trimestre de la gestation, le testicule poursuit sa descente aux enfers. Le trajet des
vaisseaux spermatiques et du canal déférent témoigne de ce trajet. L’enveloppe
péritonéale étirée forme un canal qui doit se refermer. La continuité entre les
orifices profond et superficiel, ainsi créée, facilite la perméabilité du canal
inguinal à un engagement de viscères glissant sur la membrane péritonéale dès
que la pression intrapéritonéale le permet.
Quand l’enfant respire, il crée un gradient de
pression qui empêche la fermeture du sac herniaire. Comme le péritoine sécrète
, la synéchie spontanée n’existe pas. Par contre, si l’orifice est petit, le
contenu peut ne s’engager qu’à l’adolescence lors d’un sport ou chez l’adulte
en atrophie musculaire. La survenue brutale d’une voussure chez l’homme jeune
correspond en fait souvent à une hernie inguinale indirecte congénitale. Une
anamnèse familiale soignée confirme souvent les antécédents.
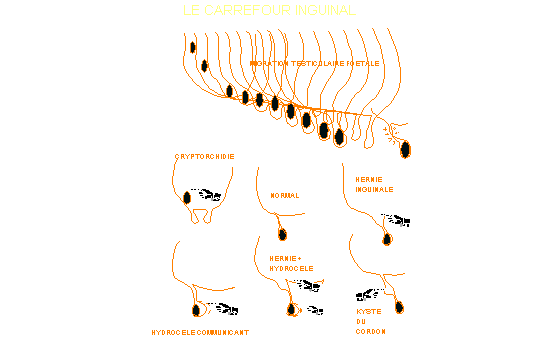
Tableau
clinique
|
Souvent diagnostiquée vers 2-3 mois ou à être
bilatérale, particulièrement lorsqu’elle |
un an au début de la marche, la hernie peut- est gauche chez le garçon. |
|
La voussure déforme le pli abdominal inférieur
et peut atteindre le scrotum. Elle peut être douloureuse et induire une
constipation. Le signe du papier de soie est caractéristique pour la différencier de l’hydrocèle. La transillumination n’est pas fiable. la manoeuvre de réduction peut être positive si l’enfant reste calme. Par contre, l’échographie peut souvent confirmer la présence de liquide dans la bourse. Garçon de 6
mois. Masse inguinale dure irréductible , gêne et cris à la palpation, pas de
signe digestif, Temp.38°
D: Hernie
inguinale droite incarcérée |
|

TABLEAU 12 LE
CARREFOUR INGUINAL: DIAGNOSTICS
|
|
|
|
|
|
|
|
|
|
|
DOULEUR |
FIEVRE |
PLI
ING. |
PL.DIGEST |
ECHO |
A.S.P. |
REFLEXE CREMAST. |
|
La
hernie inguinale |
+ |
- |
déformé |
rares |
perf |
perf |
+ |
|
La
hernie inguinale étranglée |
+++ |
+ |
déformé |
fréquentes |
perf |
perf |
+ |
|
l’hydrocèle
communicant |
+ |
- |
|
- |
perf |
- |
+ |
|
le
kyste du cordon |
- |
- |
déformé |
- |
perf |
- |
+ |
|
l’orchite
|
+++ |
+ |
|
- |
|
- |
- |
|
la
torsion testiculaire |
+++ |
+ |
|
- |
perf |
- |
- |
|
le
varicocèle |
+ |
- |
|
- |
perf |
- |
+ |
|
l’adénite
inguinale |
+ |
+ |
|
- |
perf |
- |
+ |
Ce tableau
clinique offre des guide lines: il n’est pas à l’abri des exceptions
Prise
en charge thérapeutique
La hernie inguinale s’opère dés que le
diagnostic est posé mais de manière programmée en fonction de l’état général de
l’enfant et des contraintes parentales. Là aussi, tout bandage, contention ou
manipulation est dénuée de tout fondement. Inutile aussi, chez le grand enfant
de prescrire l’arrêt des performances physiques. Le traitement chirurgical
consiste à inciser le pli abdominal inférieur et réséquer le sac inguinal en
disséquant soigneusement le cordon et les viscères éventuellement engoués dans
le processus. L’exploration hétérolatérale est controversée. Le chirurgien
tiendra compte des éléments suivants:
|
|
EXPLORATION BILATERALE |
|
AGE < 2 ans |
FAVORABLE |
|
Hernie gauche |
FAVORABLE |
|
Garçon |
FAVORABLE |
|
Fille < 5 ans |
FAVORABLE |
|
Chirurgien expérimenté |
FAVORABLE |
|
Pays médicalisé |
DEFAVORABLE |
Il s’agit d’une chirurgie mineure réalisable en
chirurgie ambulatoire sans hospitalisation mais sous anesthésie générale.
Par contre, lorsque la hernie inguinale est
compliquée d’étranglement, le traitement se fait en urgence et en deux temps si
possible.
1) LA REDUCTION
L’enfant est installé calmement dans une
baignoire tiède et idéalement prémédiqué avec du diazépam (0.5 mg/Kg), de la
morphine (0.2 mg/Kg), du Nembutal ou Thalamonal.... La réduction est
bimanuelle, progressive et complète. La manoeuvre peut prendre plusieurs
minutes. Dans les mains d’un médecin entraîné, l’irréductibilité est rare. Dans
ce cas, il peut s’agir d’un étranglement datant de plus de 8 heures, d’une
incarcération ovarienne. L’indication chirurgicale est urgente en cas d’échec.
Après une réduction chez le nourrisson, il
arrive que la récidive survienne très précocement; raison pour laquelle ces
patients sont généralement admis en observation pour 24 heures.
Chez le prématuré, la hernioraphie est pratiquée
avant la sortie de l’hôpital..
1) L’INTERVENTION CHIRURGICALE
Après la résorption de l’oedème consécutif à
l’étranglement, la hernioraphie peut être proposée.
Les diagnostics
différentiels sont évoqués et détaillés dans d’autres cours.
L’HYDROCELE
COMMUNIQUANT
|
Il s’agit de l’accumulation de liquide
péritonéal (surtout en fin de journée) autour de la bourse (parfois
bilatéral). |
Garçon de 2 ans. Masse scrotale non
douloureuse non réductible. Pas de fièvre. Pas de déformation des plis. Hydrocèle communicant persistant. |
|
Il peut être aigu et en imposer pour une
hernie inguinale étranglée. Il est non réductible. L’échographie est
déterminante. La majorité des hydrocèles disparaissent spontanément avant l’âge
de 18 mois par fermeture du C.P.V. et résorption par la vaginale. En cas
d’échec, le chirurgien lie le C.P.V. et assèche ainsi la vaginale. Le mépris
au-delà
de plusieurs années conduit à l’épaississement de la vaginale qui devient
sécrétante et conduit à l’hydrocèle non communiquant, dangereux pour le
testicule et nécessitant un retournement de vaginale. |
|

LE
KYSTE DU CORDON
|
|
Garçon de 2 ans et demi. Aucune plainte. Masse
inguinale hyperéchogène non réductible. Testicule normal Kyste du cordon |
|
Aussi appelé hydrocèle suspendu, il a la même
origine si ce n’est que le canal péritonéo-vaginal est incomplet. Il faut le
réséquer. |
|

L’ORCHITE
Les signes inflammatoires et/ou infectieux
dominent mais le gonflement brutal est piégeant. L’orchite ourlienne est la
plus commune. Le traitement est médical et local.
LA
TORSION TESTICULAIRE
Très dangereuse pour la fertilité, la torsion
doit être reconnue par la douleur aiguë, localisée autour d’un testicule
remonté et impalpable tant il est douloureux. L’échographie permet d’explorer
l’artère et l’hydatide éventuellement responsable du drame.
Seule une intervention immédiate sauve le
testicule.
LE
VARICOCELE
Dilatations veineuses caractéristiques. Douleurs
chroniques. Traitement par injection d’agent sclérosant ou emboligène ou
chirurgical (ligatures).
L’ADENITE INGUINALE
Elle est largement décrite dans le chapitre
d’infectiologie pédiatrique. Il arrive qu’un praticien soit induit en erreur
par une localisation très interne . L’examen minutieux des creux inguinaux, de
la bourse ou de la vulve est pathognomonique.
VIII. LES ANOMALIES PERINEALES
Ce Chapitre ne peut être détaillé tenant compte
du caractère limité de ce cours sur le plan horaire, mais il serait curieux et
regrettable que les futurs médecins n’aient même pas entendu citer les
principales causes de consultations dans ce domaine.
L’HYPOSPADE
LES
VALVES URETHRALES
LES
DUPLICATIONS
L’ATRESIE
VAGINALE
LE
SINUS URO-GENITAL
LES
ANOMALIES UTERINES
LE
SYNDROME ADRENO-GENITAL OU PSEUDOHERMAPHRODISME FEMELLE
LE
TESTICULE FEMINISANT
L’HERMAPHRODISME
VRAI
Enfin,
LA CIRCONCISION reste l’un des motifs fréquents de consultations en chirurgie
pédiatrique.
Pour les futurs médecins intéresses à compléter
leur culture médicale dans le domaine, ils sont invités à consulter les auteurs
cités au début du syllabus et notamment Essentials
of pediatric surgery de Marc ROWE
IX. LES DOULEURS ABDOMINALES
LE L’ENFANT
La douleur abdominale est l’un des principaux
symptômes à élucider chez l’enfant.
Le TABLEAU suivant résume l’algorithme qui
répartit ce symptôme vers les grands syndromes concernés
DOULEURS
ABDOMINALES AIGUES (DAA)
Considérons que l’anamnèse et la fiabilité de
l’environnement excluent un traumatisme (roulage, sport, scolaire...), le
syndrome de l’enfant battu, et un contexte de plaintes digestives ou urinaires
récurrentes ; si le tableau date de moins de 10 jours, l’anamnèse conduite
correctement amènera le clinicien vers le diagnostic différentiel suivant.
1. Anamnèse
Si l’interrogatoire de l’enfant est souvent
assisté par les parents ( même jusque 18 ans !), il peut apporter parfois
toutes les informations dont le clinicien a besoin pour poser son premier
diagnostic.
Parmi les questions-clés, on rappelle
l’importance du moment de début des symptomes, de la chronologie des
événements, et des éléments qui aggravent ou soulagent le tableau. Au cours de
l’entretien réalisé dans le calme absolu, et tout le monde étant assis autour
de l’enfant , on évitera de recourir au terme de douleur et de souffrance pour
se limiter aux plaintes objectivées par l’enfant. Un maximum de précisions
concerne le fonctionnement digestif, la prise de médicaments, les antécédents
opératoires et hospitaliers ainsi que le comportement social et scolaire de
l’enfant.
2. Examen physique
Au cours, nous discuterons pratiquement de
l’examen de l’abdomen de l’enfant , du nourrisson de 1 MOIS à l’adolescente de
16 ANS.
Voici les principaux signes à connaître :
·
La respiration de l’enfant (inspecter avant d’ausculter)
·
La couleur des téguments
·
L’attitude de l’enfant (agitation,
instuporation, marche...)
·
L’importance ou la présence d’un péristaltisme
(ausculter avant de palper)
·
La recherche de la défense par les signes du ballon
et de superman
·
La palpation superficielle
·
La palpation profonde de l’hypochondre gauche
vers la fosse iliaque droite
·
Ne pas oublier les creux inguinaux
La prise des paramètres comme la température
rectale ne se fait pas avant d’examiner l’abdomen !
Pendant l’examen, on s’adresse à l’enfant et pas
à ses voisins !
On n’oublie pas que l’enfant à le droit à sa
pudeur ; on diffère le toucher rectal s’il n’est pas urgent ou nécessaire.
Les indications de celui-ci seront présentées au cours.
3. Les examens complémentaires.
Ils sont demandés progressivement en fonction
des éléments cliniques ;
La biologie doit comporter le CRP, les
transaminases, un comptage leucocytaire (contributif avec beaucoup de
prudence), un PTT.
L’examen des urines est systématique
La radiographie de l’abdomen sans préparation
(ASP) doit être faite et lue.
L’échographie est
fréquemment demandée mais nécessite la perfusion si l’on veut maintenir
l’enfant à jeûn.
4.
La
stratégie diagnostique.
Elle dépend avant tout de l’âge.
< 3 MOIS
coliques
du nourrisson
reflux
gastro-oesophagien
sténose
du pylore
hernie étranglée ou incarcérée
torsion
testiculaire
malrotation - sténose duodenale - volvulus
TABLEAUX
d’aide au D.D.


3
MOIS - 1 AN
A cet âge, le contact devient plus aisé avec
l’apparition d’une compréhension plus grande. Par contre, l’enfant est plus
méfiant, plus costaud et le calme de l’examen joue un rôle fondamental.
TABLEAUX d’aide au D.D


1
AN - 3 ANS
TABLEAUX d’aide au D.D


3
- 11 ANS
En dehors des diagnostics rares, comme la
pancréatite, le purpura d’Hénoch-Schönlein, d’autres étiologies sont fréquentes
et difficilement quantifiables (intolérance ou intoxication alimentaire) ;
ces dernières sont régulièrement gérées par la famille elle-même.
TABLEAUX d’aide au D.D


>
11 ANS
Avec la croissance et l’âge, la variété des
diagnostics augmente considérablement. Progressivement, de multiples maladies
de l’adulte touchent ces jeunes patients. en revanche , il devient
possible de recourir aux examens d’imagerie qui étaient évités chez l’enfant
(CT scanner, RMN, ...)

QUELQUES
REGLES D’OR

En
conclusion, la mise au point d’une douleur abdominale chez
l’enfant requiert beaucoup d’expérience et de sagacité. L’abord
médico-chirurgical est profitable. De plus en plus, l’échographie abdominale
contribue au diagnostic chez le jeune enfant. Dans les cas complexes, on
n’hésite pas à demander un CT scanner ou tout autre examen plus sophistiqué ou
irradiant.
X.
LA PROCTOLOGIE COURANTE
La proctologie pédiatrique commence avec les
malformations congénitales de la région ano-rectale. Celles-ci sont traitées en
milieu hospitalier même si tout omnipraticien doit pouvoir les discerner .
A côté de ces dernières, plusieurs maladies
anales sont fréquemment rencontrées en pratique quotidienne.
Nous évoquerons les diagnostics suivants :
·
la fissure anale et la constipation
·
les fistules ano-rectales
·
le prolapsus rectal
A. LA FISSURE ANALE ET LA CONSTIPATION
La
fissure anale est la cause la plus classique d’hémorragie rectale chez
l’enfant. Il s’agit d’une déchirure superficielle du canal anal souvent
produite par l’exonération d’une selle dure dans le cadre d’une constipation.
Douloureuse, elle entraîne un réflexe antalgique de contraction du SAI et une
augmentation de la squeeze pressure. L’enfant évite l’exonération et engage un
cercle vicieux puisque chaque selle dure va rouvrir la plaie. Les selles
peuvent être tachées de sang rouge. La fissure anale peut être associée à une
maladie de Crohn ou à une leucémie.
L’examen
de l’anus confirme l’anamnèse ; le traitement consiste à imposer un régime
laxatif pendant plusieurs semaines ; les symptômes disparaissent
lentement.
B.
LES FISTULES ANO-RECTALES
Elles
sont souvent associées à un passé d’abcès ano-rectal, comme chez l’adulte.
Souvent
avant un an, un abcès cryptique peut survenir. Il se draine parfois
spontanément à la peau ou après drainage filiforme chirurgical . Après un
traitement antibiotique, il peut être nécessaire de réaliser une fistulectomie
chirurgicale. Ici aussi, il faut se méfier de la première manifestation d’une
maladie de Crohn.
C. LE PROLAPSUS RECTAL
Fréquent chez l’enfant handicapé mental grave,
il peut survenir sur une constipation sévère du petit enfant. Il peut être
muqueux ou complet. Survenant entre 2 et 3 ans, il est souvent idiopathique
mais doit faire penser à une mucoviscidose ou à une rectocolite
ulcéro-hémorragique. Les parents rapportent souvent des épisodes d’efforts
défécatoires importants.
Si le traitement médical peut suffir, il existe
des alternatives chirurgicales pour les formes graves évolutives : la
sclérose sous-muqueuse, l’électrocoagulation rétrorectale ou le méchage
rétrorectal.
XI.
LES MASSES ABDOMINALES ET THORACIQUES
Ce chapitre sera développé de manière
multidisciplinaire selon les possibilités horaires. Une clinique lui est
destinée.
Pour l’étudiant qui souhaite approfondir , il
trouvera quelques aperçus dans l’atlas de Liebert (voir réf .)
XII. LES TUMEFACTIONS
CERVICO-FACIALES
La sphère cervico-faciale de l’enfant est le
siège d’une multitude de pathologies acquises ou congénitales qui concernent la
pratique de tout clinicien confronté aux enfants en bas-âge.
Nous nous limiterons aux diagnostics les plus
fréquemment rencontrés en pratique générale et aux urgences de tout hôpital
ou polyclinique.
Les malformations labio-palatines sont
spécifiquement et immédiatement référées aux chirurgiens plasticiens
pédiatriques. Elles seront étudiées dans le cours de chirurgie plastique et
reconstructrice.
Nous passerons en revue plusieurs pathologies
qui seront illustrées au cours dans le but de faciliter le diagnostic
différentiel parfois délicat.
LES INCLUSIONS
DERMOIDES.
Lorsque les ébauches ectodermiques fusionnent
sur la ligne médiane, des résidus ectodermiques pré-différenciés peuvent être
inclus sous la peau et évoluer vers des kystes. Ces formations circulaires
inesthétiques sont souvent sus-orbitaires ou nasales. Elles peuvent mimer un
kyste thyréoglosse sur la ligne médio-cervicale. L’exérèse chirurgicale est
difficile mais toujours demandée. L’affection ne se complique pas; les
dégénérescences ne sont pas décrites.
LES RESIDUS
BRANCHIAUX
à la quatrième semaine du développement
embryonnaire, des bourgeons latéraux et symétriques apparaissent de part et
d’autre du massif cervico-facial.
Les
invaginations ectodermiques correspondent chacune à une fente ou arc branchial.
A chaque niveau correspond une différenciation neuro-ectodermique et
mésodermique spécifique.
La présentation clinique des résidus branchiaux
varie de la forme kystique qui peut s’abcéder à la forme fistulisée. On
observe, alors, un écoulement chronique. L’affection n’est pas douloureuse en
l’absence d’inflammation.
L’examen
comprend l’inspection de la cavité buccale et la palpation soignée de la région
cervico-faciale dans son entièreté.
L’échographie est contributive surtout dans les
formes enkystées.
Le traitement chirurgical est proposé en dehors
d’un épisode septique , traité, lui, par une antibiothérapie à large spectre.
La résection se fait par de multiples petites incisions après injection de bleu
de méthylène.
Le packing pharyngé permet de reconnaître la
communication des résidus de la IIème fente avec la fosse amygdalienne. L’intervention
doit s’étendre à la membrane sous peine de récidive.
|
Ier
arc |
Conduit
auditif externe, oreille moyenne, mastoide |
|
|
Iième
arc |
Palais,
amygdale |
Résidu
le plus fréquent |
|
IIIème
arc |
Parathyroide
sup. |
|
|
Ivème
arc |
Thymus,
Parathyroide inf. |
|
|
... |
... |
|
|
|
|
|
LOCALISATION
DES RESIDUS BRANCHIAUX
LE KYSTE
THYREOGLOSSE
Il s’agit d’un résidu kystique médian situé
entre la base de la langue et la glande thyroïde. La symptomatologie ne
survient qu’après quelques mois ou années. Contrairement au kyste dermoïde, le
kyste thyréoglosse est solidaire de l’os hyoïde. Si la glande thyroïde n’est
pas clairement palpable, on conseille de réaliser une scintigraphie
thyroïdienne.
Le traitement est chirurgical. En dehors d’un
éventuel épisode infectieux traité par antibiothérapie, la résection complète
doit être réalisée, emportant l’isthme hyoïdien sous peine de récidive.
LE LYMPHANGIOME
CERVICAL
Comme tout lymphangiome (hygroma kystique), la
forme cervicale est une anomalie congénitale du réseau lymphatique conduisant à
l’accumulation de lymphe au sein de macrokystes infiltrant progressivement le
tissu conjonctif sous-cutané et les tissus profonds. Malgré le caractère
infiltrant (épargnant les structures neuro-vasculaires auxquelles il adhère
fortement), le lymphangiome cervical reste bénin. Les pires récidives
loco-régionales ne donnent jamais de métastases à distance.
La présentation cervicale est spectaculaire
surtout si elle est néonatale. La tumeur est translucide, rénitente mais rapidement
sous tension. Si elle saigne, elle apparaît bleutée. Les formes graves
obstruent les voies aériennes supérieures et l’oropharynx.
Le diagnostic peut être confirmé par échographie
et parfois par Ctscanner. Il faut éviter les ponctions dangereuses à maints
égards. Une infection iatrogène ou une hémorragie peuvent survenir.
Le traitement est chirurgical et consiste en une
exérèse complète en un ou plusieurs temps. Un stimulateur électrique guide le
chirurgien dans la dissection fine des nerfs faciaux inclus dans le processus.
Une trachéotomie peut être nécessaire.
Les formes axillaires, thoraciques, abdominales
présentent les mêmes caractéristiques mais leur traduction propédeutique est
différente. Les membres sont moins souvent atteints. Les réinterventions sont
fréquentes et demandent une expertise chirurgicale approfondie de la maladie et
de l’anatomie opératoire.
LES
DYSEMBRYOMES PRE-AURICULAIRES
Devant le tragus, indépendant d’une fente
branchiale, des nodules, sinus ou kystes peuvent être observés. Ces
dysembryomes sont complexes, adhèrent fermement au tissu sous-cutané et peuvent
contenir un cartilage distinct ou relié
au cartilage auriculaire. Propices à la surinfection et inesthétiques (les
petites filles atteintes veulent systématiquement porter les cheveux courts),
ils font l’objet d’une exérèse élective ; attention ! si la tumeur
est petite, son exérèse n’en demeure pas aisée.
Il n’y a pas de récidive.
LA RANULA
Il s’agit d’un kyste muqueux sous-lingual
occupant le plancher buccal. L’hypothèse la plus admise est une obstruction
d’un canal salivaire sous-lingual. La ranula n’est pas douloureuse ; le
kyste est revêtu d’un épithélium et le liquide contient des amylases
salivaires. La marsupialisation du kyste guérit généralement l ‘enfant.
LE TORTICOLIS
CONGENITAL
Un enfant de quelques semaines ou de quelques
mois se présentant avec une rotation systématique et une inclinaison inverse de
la tête est suspect de torticolis congénital. Après un accouchement dystocique,
l’élongation ou la rupture du sterno-cleïdo-mastoïdien cicatrise de manière
fibreuse. Une rétraction et le raccourcissement conduisent à une position qui
associe la rotation de la tête vers le côté opposé à la lésion et une
inclinaison homolatérale. Le diagnostic est clinique (masse dure sur le trajet
du SCM et non réductibilité de la malposition. Un examen ultrasonique confirme
le diagnostic. Mésestimer ce diagnostic condamne le traitement physiothérapique
qui guérit par la manipulation progressive 75 % des cas. Dans les formes ignorées,
on assiste à une atrophie du trapèze et à une fixation définitive de la
position. Seule une chirurgie correctrice peut corriger le problème. Il n’y a
pas de consensus mais la section chirurgicale suivie d’une kinésithérapie
prolongée est la technique la plus simple.
LA PAROTIDITE
La parotidite ourlienne fait partie des
diagnostics différentiels classiques des masses cervicales. Souvent unilatérale
au début et brutale, elle peut être confondue avec d’autres masses ou tumeurs.
Le point-clé est la douleur ; en outre des signes généraux l’accompagne
souvent.
LES
ADENOPATHIES
Les adénopathies cervicales font l’objet d’une
vaste matière pour le pédiatre.
Elles constituent, bien évidemment la cause la
plus fréquente des tuméfactions cervicales chez le jeune enfant.
Attitude
L’examen recherche les caractéristiques locales
(localisation, mobilité, inflammation, douleur, rénitence,...), les signes
régionaux (otite, amygdalite, rhinite, problème dentaire,...) et puis les
plaintes générales (fièvre, asthénie, irritabilité, inappétence, ...)
Diagnostic
différentiel
Adénite pyogène
Adénite
tuberculeuse
Adénite virale
Toxoplasmose
Adénite à
mycobactéries
Lymphoréticulose
bénigne ( griffe du chat)
Raretés
Examens
complémentaires
La biologie, une virologie, une radiographie de
thorax, un frottis de gorge sont les examens de première ligne. Viennent
ensuite l’intradermo-réaction, les hémocultures, l’échographie cervicale.
Sanction
chirurgicale
Si la mise au point oriente vers une affection
non infectieuse, une tuberculose ganglionnaire ou une abcédation, un acte
chirurgical peut être indispensable.
Parmi les techniques, on citera :
1. La
biopsie-exérèse à la recherche d’un cancer des tissus mous
2. La
biopsie-frottis à la recherche d’un lymphome
3. La
ponction-exérèse à la recherche d’une tuberculose
4. Le
drainage filiforme en cas d’abcès purulent.
Les adénites cervicales d’installation lente et
progressive peuvent être confondues avec une tuméfaction congénitale telle
qu’un résidu branchial. Il faut donc insister pour que pédiatres, généralistes
et chirurgiens gardent tous les diagnostics en tête, même ceux des pathologies
qu’ils voient rarement ou exceptionnellement.
LE TERATOME CERVICAL
Les tératomes à cellules germinales sont bénins
mais infiltrent de manière spectaculaire les tissus mous, comprimant les voies
respiratoires supérieures. Les résections peuvent donner d’excellents
résultats.
|
Nouveau-né,
masse cervicale dure et fixée. Hétérogénéité ultrasonique sans liquide. Pas
de douleur, aucun signe biologique. D: Teratome cervical |
|
|

XIII. LA TRAUMATOLOGIE
COURANTE
Le squelette de l'enfant se caractérise par sa
relative souplesse, son pouvoir de consolidation, ses cartilages de
conjugaison, et ses épiphyses non ossifiées.
La présence de traumatismes osseux répétés, de
l'association entre fractures, brûlures et autres sévices doit faire suspecter
le syndrome de l'enfant battu
(incidence : 300 / 106 X AN)
L'étiologie et la localisation des fractures
sont particulières
chez l'enfant.
On ne retrouve pas les traumatismes osseux classiques
de l'adulte:
A. LA
MALTRAITANCE
SYNDROME DE L’ENFANT BATTU
Définition:
Agression physique, mentale, sexuelle, ou négligence des soins nécessaires
conduisant à une menace ou une atteinte de sa santé ou de son bien-être.
L’anamnèse et
le comportement parental sont parfois évocateurs mais aussi quelquefois
remarquablement masqués.
L'enfant est
craintif, taiseux, irritable ou instuporé, souvent sale et
négligé.On note
souvent en dessous de 4 ans:
· hématomes périorbitaires
· brûlures de cigarettes
· morsures
· hématomes (boucles de ceintures)
· hématomes sous-duraux
· malnutrition
les facteurs
prédisposants:
· parents battus
· mère de moins de 20 ans
· éthylisme
· chômage
. séparation et divorce
· handicap mental ou physique
DISPOSITIONS A
PRENDRE:
· hospitaliser l'enfant
· mise de la famille sous contrôle
socio-médical
· traiter l'enfant
· prise en charge pluridisciplinaire
Ne
pas agresser les parents, culpabiliser
Ne faire
intervenir le pouvoir judiciaire ou la police qu'en cas
de danger
vital imminent.
B. LES FRACTURES FREQUENTES et TYPIQUES
LA PRONATION
DOULOUREUSE
LE DECOLLEMENT
EPIPHYSAIRE DU RADIUS
LA MOTTE DE
BEURRE DE L'AVANT-BRAS ET LA # DE
MONTEGGIA
LES # DE LA PALETTE HUMERALE
LA # (OUVERTE) DU TIBIA
LA # DIAPHYSAIRE DU FEMUR
LA # DE LA CLAVICULE
par opposition:
LES # TYPIQUES DE L'ADULTE
LA # DU COL
FEMORAL
LES # COSTALES
LES # DE LA
CHEVILLE
LES LUXATIONS
LA # DU COL DE
L'HUMERUS
.............
LA PRONATION
DOULOUREUSE
Le tableau
synoptique reprend les points-clés. L’affection est fréquente, bénigne et doit
faire l’objet d’une reconnaissance rapide et de la réduction précoce.

La pronation
douloureuse :
|
Le bras est
en pronation antalgique.
L’opérateur saisit la main de l’enfant avec sa main homolatérale et prend
gentiment le coude avec la main opposée |
Tout en
maintenant une pression des doigts sur l’articulation huméro-radiale,
l’avant-bras est amené lentement mais fermement en supination |
Une flexion
de l’avant-bras sur le bras termine la manoeuvre et achève la réduction à
travers le ligament |
|
|
|
|

LE DECOLLEMENT
EPIPHYSAIRE DU RADIUS
La fracture
typique de Pouteau-Colles n’existe pratiquement pas chez l’enfant. Elle est
remplacée par le décollement au niveau du cartilage de conjugaison. La
caractéristique est que la radiographie peut-être négative ou peu contributive.
Le diagnostic est clinique ; il existe une douleur exquise au niveau de ce
cartilage. L’immobilisation est de rigueur pendant au moins 4 mois.
|
|
PRESENTATION Chute à vélo ou d'un arbre (4 - 10 ANS) douloureuse DD diagnostic
d'abord clinique se
méfier de la # du coude RX Glissement
épiphysaire ou
presque aucun déplacement ATTITUDE Réduction sous narcose puis si
doute scintigraphie |
|
|
|
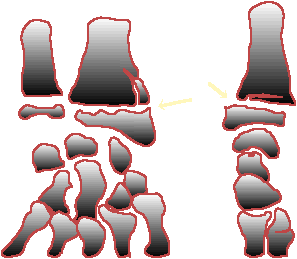
LA MOTTE DE
BEURRE DE L'AVANT-BRAS ET LA # DE
MONTEGGIA

LES # DE LA PALETTE HUMERALE
Elles
constituent le piège même de la traumatologie pédiatrique.
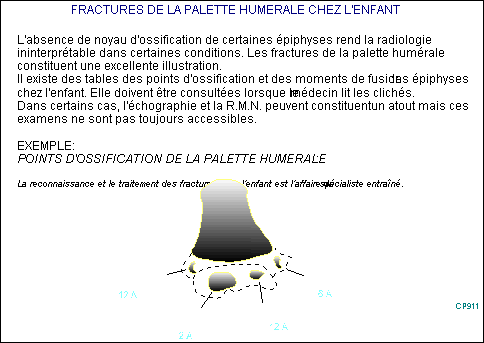
LA # (OUVERTE) DU TIBIA
Le diagnostic
est cliniquement évident. Il requiert un transport médicalisé et une traction
rapide. L’immobilisation peut se faire par fixation externe.
LA # DIAPHYSAIRE DU FEMUR
Le diagnostic
est aisé ; l’immobilisation se fait par traction ou fixation externe.
L’enclouage et les fixations internes sont abandonnées.
LA # DE LA CLAVICULE
Fracture d’été
classique, elle survient souvent au cours d’une chute à vélo ou en roller. Le
diagnostic est clinique et l’immobilisation se fait par un bandage
bi-scapulaire en huit. Les indications opératoires sont très rares.
XII. ORTHOPEDIE
COURANTE
Les
malformations ou malpositions congénitales affectant le système
musculo-squelettique sont nombreuses en pratique quotidienne. Elles peuvent
être stables ou évolutives. On distingue des malformations congénitales
d’étiologie génétique ou tératologique des déformations posturales et statiques
acquises.
|
|
Affections
congénitales |
Affections
posturales acquises secondaires |
|
Générales |
Arthrogrypose Syndrome
de Down Syndrome
de Marfan Osteogenesis
imperfecta Dysplasie
squelettique |
Affections
neurovégétatives Maladies
neuromusculaires Affections
métaboliques Rachitisme |
|
Focales |
Pied
bot varus équin Luxation
congénitale hanche Méromélie Asymétrie
des membres inférieurs |
Torticolis
congénital (trauma) Poliomyélite
(infectieux) Metatarsus
adductus Scoliose Genu
valgum Pes
planus - cavus |
Exemples d’affections ostéo-articulaires
(classification de Mac Mahon modifiée).
Toutes
ces déformations squelettiques (ostéo-articulaires) sont d’apparition
progressive, évolutives et indolores. Il s’agit d’enfants présentés en
consultation élective ou en maternité.
Par
opposition, on distingue les maladies aiguës ou subaiguës acquises,
douloureuses, accompagnées de symptômes généraux ou d’impotence fonctionnelle
brutale qui conduisent l’enfant à la garde d’un hôpital ou en consultation
urgente, voire en appel à domicile.
Il
est bien entendu illusoire de vouloir brosser un panorama complet des
affections orthopédiques pédiatriques. Afin de donner un aperçu de quelques
problèmes courants spécifiques à l’enfant, seront abordés dans le présent
chapitre :
A.
Le pied bot et ses variantes
B.
La luxation congénitale de hanche
C.
Les causes de boiterie
D.
Scoliose et attitude scoliotique
E.
L’ostéomyélite
A. Le
pied bot et ses variantes
Le
pied bot varus équin est connu et décrit depuis l’Antiquité. Il s’agit d’une
affection relativement fréquente (±
250 nouveaux cas par an en Belgique).
Comme
le nom l’indique (talipes equino cavo varus), il s’agit d’une malformation
complexe de l’arrière et de l’avant-pied consistante en un équin, un métatarsus
adductus et un varus de l’arrière-pied avec subluxation calcanéo-astragalienne.
L’étiopathogénie est complexe puisqu’une prédisposition génétique a été décrite
dans certaines familles; d’autre part le pied bot varus équin peut être
reproduit au laboratoire par la création d’un oligohydramnios et du syndrome
amniotique. En outre, les formes malpositionnelles non fixées réductibles en
quelques jours ou en quelques semaines plaident pour une compression mécanique
intra-utérine.
Quels
que soient les facteurs étiopathogéniques possibles, les formes fixées non
réductibles doivent être traitées précocement. La reconnaissance du pied bot
est devenue systématique dans les pays occidentaux. Elle reste mésestimée dans
les contrées moins développées médicalement (Afrique Centrale, Extrême-Orient
ou Amérique du Sud).
Mise au point
Le
diagnostic est bien entendu aisé sur le plan clinique mais l’échographie et les
radiographies du pied permettent d’apprécier les angulations notamment avant le
traitement et d’établir un premier pronostic concernant l’efficacité d’un
traitement orthopédique non invasif ou la nécessité du recours à une correction
chirurgicale.
La
forme complète doit être distinguée du métatarsus adductus simple et du pied
calcanéo valgus.
Traitement
50%
des enfants peuvent être traités par plâtre et attelle.
Le
traitement orthopédique commence dès le premier jour après la naissance ou dès
le diagnostic. La mise en place de taping précoce permet la correction rapide
des formes malpositionnelles. Le stretching et la kinésithérapie jouent un rôle
dans les formes touchant essentiellement les tissus mous.
La réduction orthopédique du pied bot doit
être progressive. Il a été démontré que l’agressivité de la réduction
provoquait des lésions des cartilages articulaires et des raideurs articulaires
impossible à récupérer ultérieurement. Le recours à des attelles en rotation
externe de type Denis Browne ou assimilées peut être prolongé pendant plusieurs
mois notamment la nuit.
Le
traitement orthopédique est donc de règle pendant les 6 premiers mois.
Après
ce stade et selon les écoles, une chirurgie des tissus mous peut être
envisagée. Celle-ci consiste en la libération de l’appareil ligamentaire et à
d’éventuels allongements tendineux permettant la correction complète.
Le
maintien en position de réduction pendant plusieurs mois s’avère indispensable
après l’intervention (intervention de Turcot). Plus tard des interventions
correctrices complémentaires au niveau osseux peuvent être réalisées dans les
cas réfractaires. Elles peuvent conduire dans certains cas rares à l’arthrodèse
de certaines articulations.

Le
pronostic peut être considéré comme excellent dans 85 à 90% des cas.
Le
métatarsus adductus est une forme mineure. Le métatarse et les orteils sont en
adduction par rapport à l’arrière-pied. La réduction et le maintien en position
correcte sous plâtre permettent presque toujours la réduction et le traitement
définitif.
B. La
luxation congenitale de hanche
La
luxation congénitale de hanche regroupe un éventail d’anomalies coxofémorales
s’étendant de la dysplasie acétabulaire diagnostiquée à l’âge de 20 ans jusqu’à
la luxation intra-utérine complète de la tête fémorale dans l’acétabulum
présent à la naissance.
Sur
le plan clinique, l’omnipraticien et le pédiatre distingueront la luxabilité de
hanche révélée par le signe d’Ortolani (1000 cas annuels en Belgique) de la
luxation congénitale avérée (- 300 cas en Belgique).
Des
facteurs hormonaux maternels à l’incidence familiale de multiples facteurs
étiopathogéniques ont été démontrés. Une fois de plus, l'oligohydramnios
expérimental ou clinique joue un rôle.
L’histoire
naturelle de la luxation non diagnostiquée conduit à une boiterie dès la marche
et à une arthrose coxofémorale dégénérative conduisant à une impotence
fonctionnelle évolutive.
Signes cliniques et
dépistage
Le
signe d’Ortolani est recherché systématiquement lors de tout examen du
nouveau-né. L’enfant est couché sur sa table à langer. Le médecin saisit les
genoux en apposant le pouce sur la face interne du condyle fémoral interne et
l’index le long du bord externe de la cuisse. Le fémur est amené à 90° et
gentiment en abduction. En palpant le grand trochanter à l’aide du majeur, les
mouvements de chaque fémur dans les cavités acétabulaires peuvent déclencher un
clic caractéristique qui peut être uni- ou bilatéral.
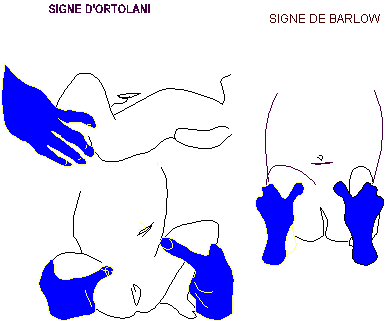
Le
chirurgien et l’orthopédiste pédiatrique préfèrent souvent au test d’Ortolani
le test de Barlow. Celui-ci consiste à stabiliser d’une main le bassin par une
prise de celui-ci au niveau du pubis et du sacrum à l’aide du pouce et du
majeur. L’autre main induit une abduction et une rotation interne de
l’articulation coxofémorale attestée. La subluxation est alors aisément
perceptible. Le signe de Barlow permet mieux de tester chaque hanche
alternativement et permet de mieux dépister les formes bilatérales.
La
luxation de la hanche proprement dite présente à la naissance peut être
observée simplement par l’examen clinique des plis inguinaux et fessiers. Une
asymétrie du pli fessier et l’impossibilité d’une abduction en décubitus
ventral signent le diagnostic.
L’examen
clinique des hanches chez l’enfant doit être réalisé plusieurs fois pendant les
premières semaines si possible lorsque l’enfant est calme et relâché.
Le
diagnostic peut être complété par l’échographie et par l’examen radiologique.
L’examen radiologique ne peut démontrer avec précision la position des têtes
fémorales puisque celles-ci ne sont pas ossifiées à la naissance. Le cartilage
épiphysaire est par contre parfaitement visualisé à l’échographie. La dysplasie
acétabulaire est mesurable sur les clichés radiologiques standards.
Traitement
Le
premier traitement simple consiste à maintenir l’enfant en abduction.
L’utilisation de 2 langes fermés dans le dos de l’enfant permet déjà de
maintenir une abduction proche des 90° en cas d’une suspicion d’une luxabilité.
Si après quelques jours le signe persiste, le recours à des attelles plus
sophistiquées (Pavlick, Frejka ...) s’avère nécessaire. Le traitement de la
luxation persistante nécessite une traction au zénith. Les cas révélés plus
tardivement peuvent conduire à une hospitalisation et à une traction prolongée
suivie d’un traitement chirurgical plus complexe.
Une
plastie acétabulaire peut être nécessaire chez le plus grand enfant (opération
de Salter).
C. Affections
orthopediques acquises entraînant une boiterie
La
boiterie constitue un symptôme fréquent et peu spécifique en consultation de
médecine générale.
De
l’appendicite aiguë à la chaussure mal ressemelée jusqu’à la maladie de
Legg-Calvé-Perthes en passant par l’enfant battu, les diagnostics différentiels
de la boiterie sont complexes et variés. En cas de boiterie l’anamnèse et
l’examen clinique doivent orienter le médecin avant la demande d’examens
sophistiqués et d’explorations invasives.
Après
un interrogatoire des parents et de l’enfant si son âge le permet, il est
important de faire marcher le patient dans le cabinet de consultation ou dans
le couloir sur une longueur suffisante afin de localiser le côté de la boiterie
et les désordres associés.
On
veillera à vérifier si l’enfant pose correctement le pied sur le sol, si
l’extension de la jambe sur la cuisse est complète, si une bascule importante
du bassin ou des épaules sont objectivées. Il est intéressant de voir si
l’enfant est penché vers l’avant.
La
marche doit être observée sans aucun vêtement.
Les
diagnostics relevant de la sphère abdominale seront exclus par un examen
abdominal approprié notamment au niveau des orifices inguinaux, des bourses et
de l’ensemble de la cavité péritonéale.
Un
examen approfondi des pieds et des genoux permettra d’éliminer la présence de
blessures quelquefois cachées ou mésestimées.
Lorsque
la plainte se concentre au niveau des hanches, trois diagnostics sont à évoquer
et à différencier : la hanche irritable, la maladie de Legg-Calvé-Perthes et
l’épiphysiolyse.
L’ostéoarthrite
de hanche doit bien entendu être toujours exclue (voir chapitre de
l’ostéomyélite. Dans les régions endémiques, la tuberculose de la hanche doit
également être suspectée en fonction de l’état général.
1. La synovite transitoire de hanche
Cette
entité également appelée hanche irritable ou rhume de hanche consiste en
l’apparition d’une douleur et d’une impotence fonctionnelle souvent unilatérale
chez un enfant en âge préscolaire (4 à 7 ans). Un état légèrement grippal peut
être décrit par les parents. L’enfant est en principe apyrétique et présente
une légère boiterie conduisant éventuellement vers le reflux de se lever.
L’examen
clinique en décubitus dorsal démontre une limitation des mouvements de la
hanche atteinte notamment en abduction et en rotation. L’étiopathogénie est
controversée. Une étiologie virale a été évoquée et rarement démontrée. La
présence d’un traumatisme récent est quelquefois mise en évidence.
Lorsque
l’enfant est allongé sur son lit, les plaintes régressent spontanément et la
mise en traction semble améliorer l’évolution clinique.
Diagnostic
Le
diagnostic clinique est l’absence de signes biologiques généraux, nécessite
souvent la réalisation de clichés radiographiques.
Dans
la synovite transitoire de hanche les clichés radiologiques ne peuvent montrer
tout au plus qu’une asymétrie des espaces articulaires coxofémoraux et
quelquefois un léger oedème des tissus mous.
Un
léger bombement de la capsule articulaire peut être objectivé.
La
scintigraphie osseuse au disphosphonate est négative excluant l’ostéomyélite et
la maladie de Legg-Calvé-Perthes.
Pour
certains auteurs, la synovite transitoire de hanche peut être un premier stade
réversible de la nécrose aseptique. Le passage d’un état de hanche irritable à
une maladie de Legg-Calvé-Perthes avérée a été démontré.
Traitement
La
mise au repos de l’enfant et la traction permettent le contrôle de la douleur.
L’évolution favorable après 8 ou 15 jours confirme la bénignité de l’affection.
Le pronostic est donc globalement favorable.
Dans
quelques cas la bilatéralité peut être observée.
2. Maladie de Legg-Calvé-Perthes
L’ostéochondrite
de la tête fémorale survient chez des enfants de 5 à 10 ans. Les garçons sont
plus souvent atteints que les filles.
Le
tableau clinique inaugural ressemble à la synovite transitoire de hanche mais
son évolution est beaucoup plus dramatique et conduit à la nécrose éventuellement
complète de la tête fémorale avec des séquelles considérables en cas de
non-diagnostic.
Tableau
clinique
Il
s’agit souvent d’un garçon en âge scolaire se plaignant d’une douleur au lever
avec limitation de l’abduction et de la rotation interne.
Une
légère fébrilité peut être notée mais elle n’est aucunement indispensable au
diagnostic.
L’examen
radiologique permet de définir plusieurs stades selon la classification de
Catterall.
De
la symétrie de la capsule articulaire jusqu’à la nécrose et au séquestre de
têtes fémorales, tous les stades de destruction osseuse peuvent être observées.
Le
prélèvement bactériologique est toujours négatif et l’étiopathogénie fait appel
à une dévascularisation brutale liée à la croissance épiphysaire et
métaphysaire.
Le
pronostic est d’autant plus favorable que l’enfant est jeune.
Traitement
La
mise en traction est indispensable pour des raisons antalgiques. Elle permet
également le maintien d’un espace articulaire réduisant les risques
d’aggravation de la dévascularisation de la tête fémorale.
La
notion d’une revascularisation spontanée favorise un meilleur pronostic.
L’absence de déformation complète de la structure céphalique réduit également
les séquelles.
Le
traitement chirurgical peut être proposé en cas de nécrose radiologique
évolutive. Il consiste à perforer par voie arthroscopique la tête fémorale afin
de favoriser une néo-vascularisation.
Dans
les cas de destruction importante, une chirurgie osseuse reconstructrice peut
être nécessaire. L’évolution peut conduire à une arthrose coxofémorale précoce
après quelques années.
3. L’épiphysiolyse
L’épiphysiolyse
fémorale atteint souvent les jeunes adolescents. La prédominance des sexes
dépend de l’expérience des équipes.
Tableau
clinique
La
symptomatologie est douloureuse et intermittente. L’impotence fonctionnelle est
variable.
Diagnostic
L’examen
radiologique approfondi de la hanche permet de mettre en évidence un glissement
épiphysaire externe qui peut conduire jusqu’à la subluxation.
Plusieurs
stades ont été décrits.
Prise
en charge et traitement
La
mise au repos de la hanche s’impose. Lorsque le débords de la métaphyse sur la
tête fémorale dépasse 30%, une réduction et fixation interne par embrochage
peuvent s’avérer judicieuses.
Le
pronostic à long terme est favorable.
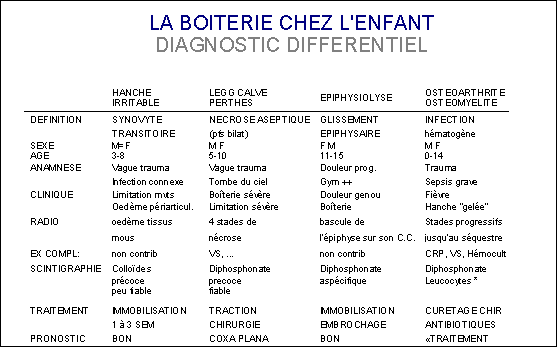
D. Scoliose
et attitude scoliotique
Les
malformations de la colonne vertébrale sont variées et fréquentes.
On
peut distinguer :
- le spina
bifida consistant à la fermeture incomplète des arcs vertébraux postérieurs
avec maintien d’une éventuelle fistule neuro-ectodermique;
- le
myéloméningocèle consistant à la non-fermeture du tube neural conduisant
éventuellement à l’extériorisation de la moelle épinière sur la ligne médiane
dorsale, - les anomalies vertébrales ou sacrées (hémivertèbre, ...);
- le
torticolis congénital consécutif à un traumatisme du sterno-cléido-mastoïdien à
la naissance;
- les
scolioses;
- attitude
scoliotique.
Si
les malformations neurovertébrales concernent essentiellement les
neurochirurgiens, l’omnipraticien et le pédiatre doivent reconnaître la
scoliose structurelle de l’attitude scoliotique secondaire.
Scoliose structurelle
Une
asymétrie des plateaux vertébraux ou des anomalies vertébrales plus complexes
peut conduire à une déformation progressive d’un segment vertébral. Une
scoliose thoracique dextroconvexe va conduire à des répercussions cervicales et
lombaires dextroconcave. La colonne évolue alors vers un double S. L’évolution
de la scoliose idiopathique est très mauvaise si la prise en charge
thérapeutique n’est pas correctement réalisée.
En
effet, lorsque la déformation devient esthétiquement visible il n’est pas rare
de mesurer une angulation de plus de 20°. On admet généralement qu’elle
s’aggrave d’1°/an. Ainsi, une angulation thoracique de 22° diagnostiquée à
l’âge de 7 ans conduit à une déformation de 45° à l’âge de 30 ans et de 90° à
l’âge de 75 ans.

Attitude scoliotique
L’attitude
scoliotique est une incurvation de la colonne vertébrale soit posturale
évoluant au cours de la croissance mais spontanément résolutive soit secondaire
et compensatoire à une anomalie des membres ou du bassin.
Le
cas le plus classique consiste en une asymétrie des membres inférieurs
entraînant une bascule du bassin et des scolioses compensatoires lombaires,
thoraciques et finalement cervicales.
L’enfant
corrige toute déformation posturale afin d’horizontaliser son regard. Une
inégalité des membres inférieurs peut être aisément mesurée par scanométrie ou
cliniquement si elle est importante.
Tableau clinique
|
|
Scoliose |
Attitude
scoliotique |
|
Diagnostic décubitus dorsal |
Pas de réduction |
Réduction |
|
Gibbosité |
+ |
- |
|
Compensation segmentaire |
Prononcée |
Faible |
|
Apophyse épineuse |
Rotation |
Pas de rotation |
|
Côtes |
Asymétrie |
Pas d’asymétrie |
|
Flexion du tronc vers l’avant |
|
¯ |
|
Position assise |
XX |
¯ |
|
Plis fessiers |
Symétrie |
Asymétrie |
Traitement
Le
traitement de l’attitude scoliotique posturale est kinésithérapique et le
pronostic est favorable. L’attitude scoliotique secondaire à une asymétrie des
membres inférieurs doit être traitée par une correction de la symétrie des
membres inférieurs (talonnette ou chirurgie d’allongement).
Le
traitement de la scoliose idiopathique évolutive est soit orthopédique
(traction, plâtre et corset) soit chirurgical (tige d’Harrington, ...).
Le
pronostic de la scoliose idiopathique avec gibbosité est souvent médiocre.
E. L’ostéomyelite
Parmi
les affections osseuses acquises de l’enfant, l’ostéomyélite aiguë hématogène
est une entité importante à connaître en pratique générale.
Sa
pathogénie, son évolution et son traitement sont à distinguer clairement de
l’ostéite classique rencontrée chez l’adulte et en général secondaire à une
contamination locale souvent traumatique.
L’ostéomyélite
aiguë hématogène est, comme le nom l’indique, une infection bactérienne aiguë
de la structure osseuse et de la cavité médullaire suite à la greffe
bactérienne amenée par voie hématogène de la métaphyse des os longs.
Dans
de nombreux territoires osseux au cours de la croissance, la vascularisation
s’avère temporairement critique et terminale.
C’est
par exemple le cas au niveau de l’épiphyse proximale du fémur et de la région
qui entoure son cartilage de conjugaison. Si le traumatisme peut constituer un
facteur déclenchant, les auteurs s’accordent sur la fréquence d’une
localisation secondaire osseuse suite à un foyer infectieux extérieur
éventuellement de la sphère ORL.
L’agent
causal dans la majorité des cas est le staphylocoque aureus (80-90%). On relève
également une incidence de streptocoque, de pneumocoque, d’Escherichia coli et d’Haemophilus
influenzae. Si l’étiopathogénie et l’analyse bactériologique du phénomène
sont convaincantes, une déficience immunitaire sous-jacente doit être
quelquefois recherchée.
L’évolution
de la greffe bactérienne métaphysaire dans la région des sinusoïdes provoque
rapidement une congestion et une hypertension au niveau articulaire si la
métaphyse est intra-articulaire ou périostée dans les autres cas.
L’hyperpression localisée induit une chute du débit vasculaire et des micro-thromboses
qui entraîne rapidement une ischémie osseuse qui conduit à la nécrose.
Le
foyer infectieux se développe en territoire nécrotique et conduit à une
destruction rapide de la structure osseuse quelquefois fort étendue.
Si
certains cas de guérison ont été décrits, l’absence de reconnaissance médicale
et de traitement médico-chirurgical approprié conduit à des séquelles
irréversibles notamment au niveau de la croissance.
Présentation clinique
On
note une altération rapide de l’état général avec fatigue, anorexie,
irritabilité, une fièvre importante atteignant quelquefois 39 ou 40° et une
douleur locale conduisant à une impotence fonctionnelle immédiate.
Le
diagnostic est d’autant plus délicat que l’enfant est en bas âge
particulièrement pendant les premiers mois de la vie.
Dans
les cas d’ostéomyélites néonatales une hypoglobulinémie est souvent rapportée.
L’examen
clinique osseux systématique de l’enfant doit être réalisé et le caractère
multifocal classique nécessite un examen très complet et journalier des membres
et des ceintures scapulaire et pelvienne.
Les
explorations biologiques révèlent une augmentation considérable de la vitesse
de sédimentation et du CRP. La leucocytose est systématiquement augmentée.
L’hémoculture permet dans 2/3 des cas une identification bactérienne rapide et
un antibiogramme précoce.
Les
clichés radiograhiques standards peuvent être difficiles à interpréter. On note
néanmoins dans la phase initiale un oedème des tissus mous, une augmentation
des espaces articulaires et un soufflage périosté qui précède ou qui est
associé à une hétérogénéité de la structure osseuse. Dans un 3ème stade, on
note la présence de lacunes avec condensation osseuse bien délimitée signant la
présence d’une nécrose installée.
La
scintigraphie osseuse au technétium ou au gallium permet une étude globale du
squelette, confirme le foyer primitif et permet d’éliminer la présence de
foyers secondaires.
Parmi
les diagnostics différentiels il faut exclure l’érysipèle, le rhumatisme
articulaire aigu et l’anémie falciforme dont les crises peuvent mimer une
ostéomyélite.
Le
diagnostic différentiel entre une ostéoarthrite et une ostéomyélite
métaphysaire est difficile notamment chez le petit enfant.
Le
dernier diagnostic à exclure est le sarcome d’Ewing dont l’allure radiologique
initiale peut mimer une ostéomyélite.
Prise en charge et
traitement
La
présence d’une fièvre aiguë avec douleurs osseuses localisées doit faire
évoquer un diagnostic d’ostéomyélite par le médecin traitant ou le pédiatre.
L’hospitalisation de l’enfant est indispensable dans les heures qui suivent
cette impression diagnostique.
Une
biologie complète, des clichés radiographiques, éventuellement une
scintigraphie confirment le diagnostic.
L’enfant
doit être immédiatement traité par antibiothérapie intraveineuse orientée vers
le pari bactériologique du staphylocoque aureus.
L’oxacilline
ou la flucocloxacilline sont les antibiotiques de choix. L’amoxycilline et les
céphalosporines peuvent également être utiles en fonction de l’antibiogramme et
de l’allergie éventuelle.
Si
les clichés radiologiques et l’évolution clinique sont défavorables, un
traitement chirurgical s’impose.
Sous
anesthésie générale, une ponction drainage percutanée du foyer d’ostéomyélite
peut être réalisée. En cas de réponse positive au niveau du prélèvement, une
incision périostée et un curetage du foyer d’ostéomyélite s’imposent.
La
décompression chirurgicale du foyer d’ostéomyélite métaphysaire doit être
rapide afin de diminuer le risque de séquestre. La réponse à l’antibiothérapie
sera d’autant plus rapide qu’une décompression du foyer d’ostéomyélite est
opérée.
L’immobilisation
antalgique est conseillée même si elle n’a pas démontré une modification dans
la durée du traitement.
L’antibiothérapie
doit être poursuivie par voie intraveineuse au-delà de 3 semaines. Dans les cas
de destructions plus importantes, elle peut être prolongée jusqu’à 6 semaines.
La
mauvaise reconnaissance d’une ostéomyélite aiguë ou son traitement inadéquat
peut conduire à une ostéomyélite multifocale ou chronique dont le traitement
s’avère alors d’autant plus complexe.
Les
prélèvements histologiques peuvent être réalisés afin d’exclure une situation
néoplasique.
L’évolution
est favorable lorsque le diagnostic est précoce et le traitement approprié.
Ce cours écrit est destiné aux étudiants en
médecine de 2ème doctorat.
Il constitue la base des connaissances
nécessaires pour réussir correctement
les
épreuves écrites ou orales à venir.
Comme tout apprentissage d’une matière clinique
par des universitaires ,
il doit
être assorti de l’acquisition de la démarche clinique, de l’accès à une
illustration des présentations classiques et d’une curiosité scientifique qui
doit mener à
la
compulsion d’un ouvrage de référence et d’une fréquentation minimale au cours ex cathedra, seule opportunité, pour le
professeur de rencontrer les étudiants
et de les faire participer à la démarche
intellectuelle.
Bon travail aux étudiants.
Jean-Jacques HOUBEN